

Deciphering the Epigenetic Code in Embryonic and Dental Pulp Stem Cells
- PMID: 28018144
- PMCID: PMC5168831
Deciphering the Epigenetic Code in Embryonic and Dental Pulp Stem Cells
Abstract
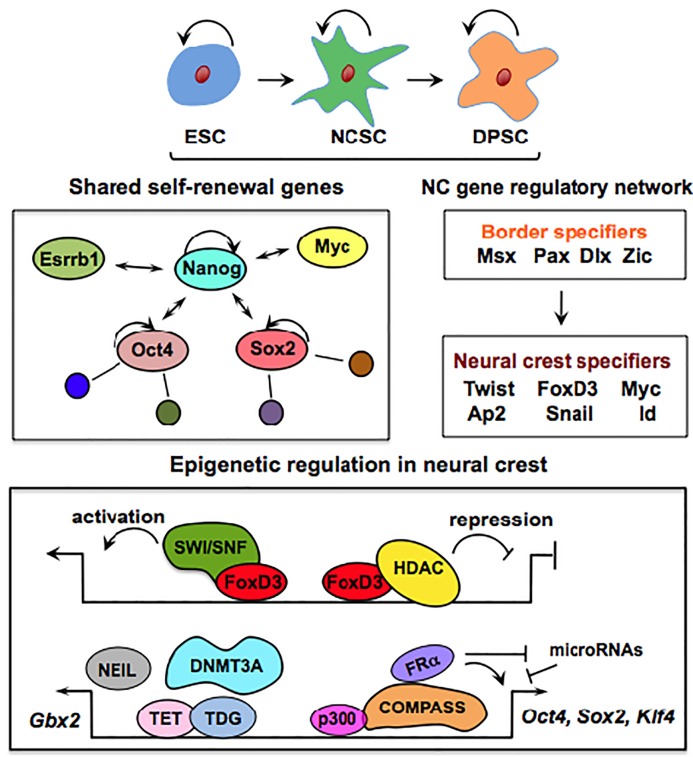
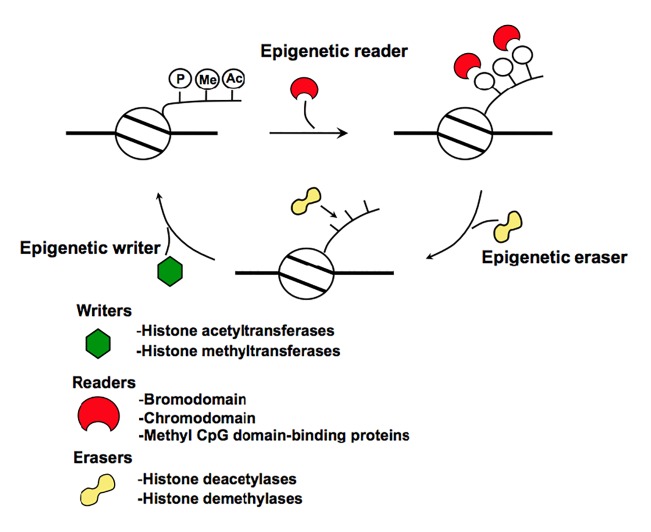
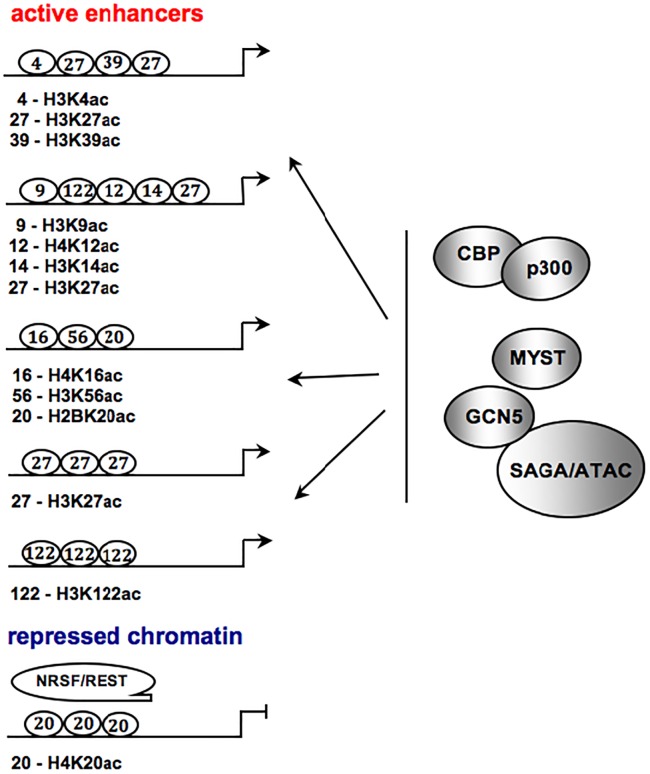
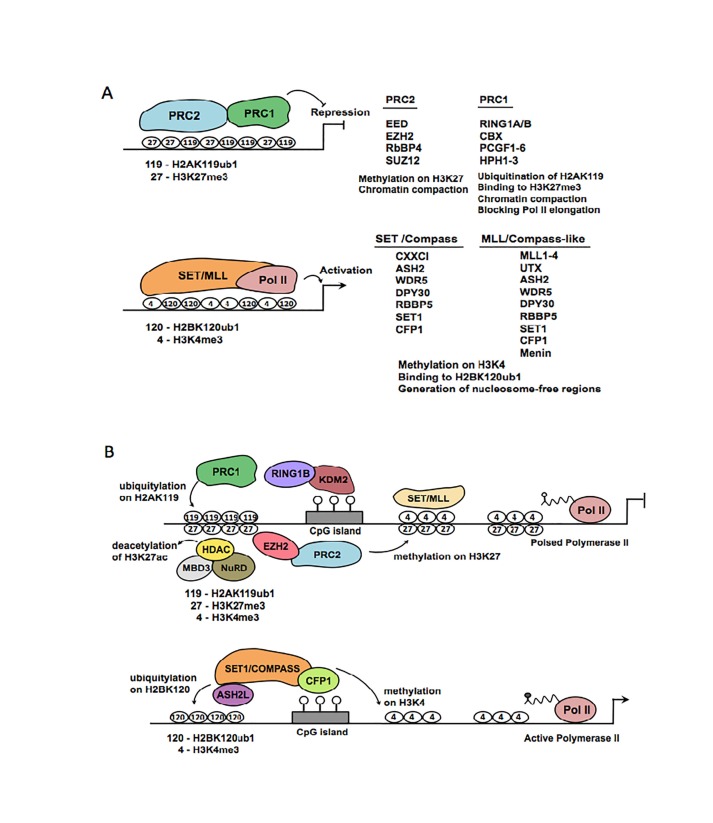
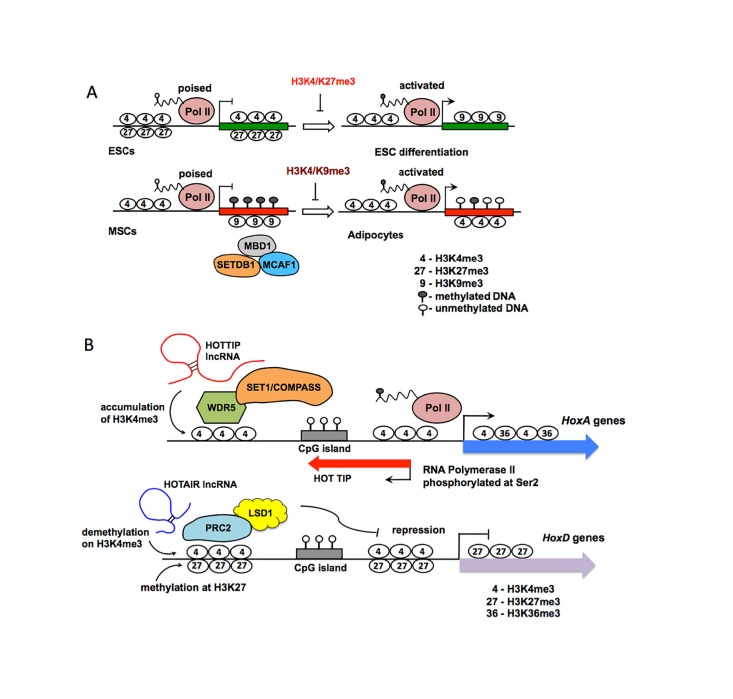
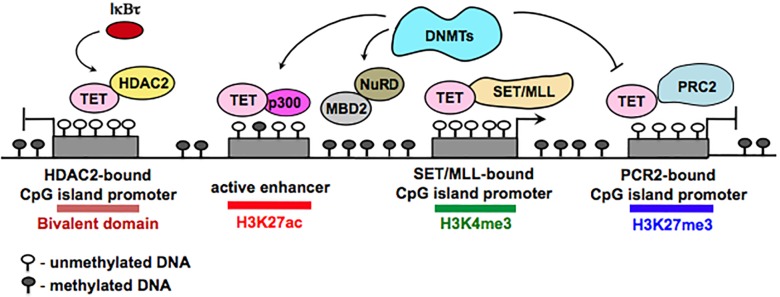
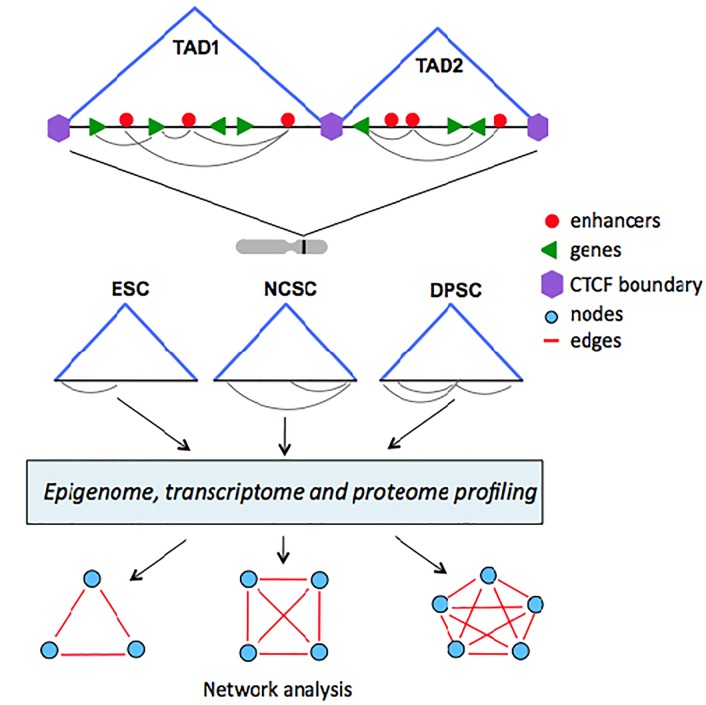
A close cooperation between chromatin states, transcriptional modulation, and epigenetic modifications is required for establishing appropriate regulatory circuits underlying self-renewal and differentiation of adult and embryonic stem cells. A growing body of research has established that the epigenome topology provides a structural framework for engaging genes in the non-random chromosomal interactions to orchestrate complex processes such as cell-matrix interactions, cell adhesion and cell migration during lineage commitment. Over the past few years, the functional dissection of the epigenetic landscape has become increasingly important for understanding gene expression dynamics in stem cells naturally found in most tissues. Adult stem cells of the human dental pulp hold great promise for tissue engineering, particularly in the skeletal and tooth regenerative medicine. It is therefore likely that progress towards pulp regeneration will have a substantial impact on the clinical research. This review summarizes the current state of knowledge regarding epigenetic cues that have evolved to regulate the pluripotent differentiation potential of embryonic stem cells and the lineage determination of developing dental pulp progenitors.
Keywords: DNA methylation; chromatin topology; dental pulp stem cells; embryonic stem cells; enhancers; histone modifications; non-coding RNA.
Figures
References
-
- Hamidi T, Singh AK, Chen T. Genetic alterations of DNA methylation machinery in human diseases. Epigenomics. 2015;7(2):247–265. - PubMed
-
- Luo Z, Lin C. Enhancer, epigenetics, and human disease. Curr Opin Genet Dev. 2016;36:27–33. - PubMed
-
- Duncan HF, Smith AJ, Fleming GJ. et al. Epigenetic modulation of dental pulp stem cells: implications for regenerative endodontics. Int Endod J. 2016;49(5):431–446. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous