

ATG16L1 governs placental infection risk and preterm birth in mice and women
- PMID: 28018968
- PMCID: PMC5161251
- DOI: 10.1172/jci.insight.86654
ATG16L1 governs placental infection risk and preterm birth in mice and women
Abstract
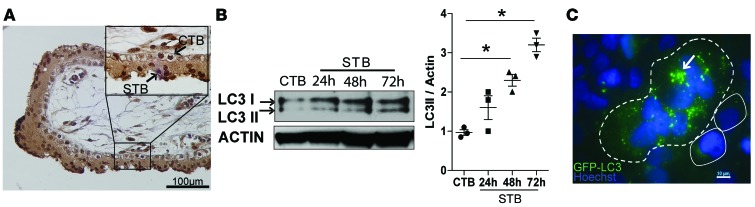
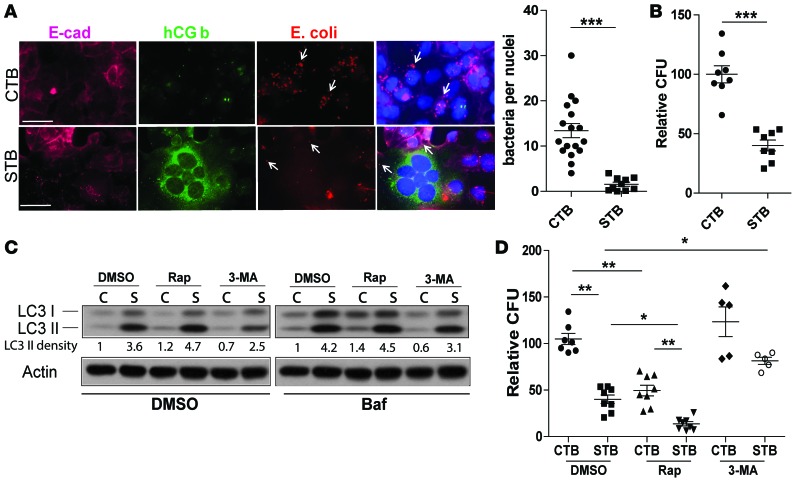
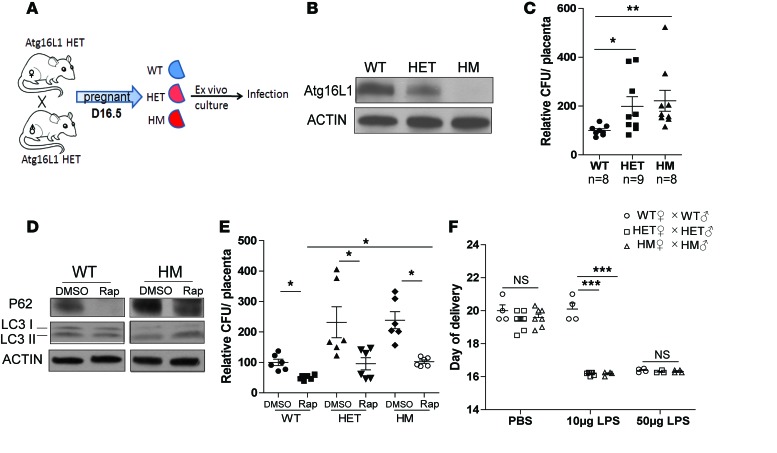
The placenta is a barrier against maternal-fetal transmission of pathogens. Placental infections can cause several adverse pregnancy outcomes, including preterm birth (PTB). Yet, we have limited knowledge regarding the mechanisms the placenta uses to control infections. Here, we show that autophagy, a cellular recycling pathway important for host defense against pathogens, and the autophagy gene Atg16L1 play a key role in placental defense and are negatively associated with PTB in pregnant women. First, we demonstrate that placentas from women who delivered preterm exhibit reduced autophagy activity and are associated with higher infection indicators. Second, we identify the cellular location of the autophagy activity as being in syncytial trophoblasts. Third, we demonstrate that higher levels of autophagy and ATG16L1 in human trophoblasts were associated with increased resistance to infection. Accordingly, loss of autophagy or ATG16L1 impaired trophoblast antibacterial defenses. Fourth, we show that Atg16l1-deficient mice gave birth prematurely upon an inflammatory stimulus and their placentas were significantly less able to withstand infection. Finally, global induction of autophagy in both mouse placentas and human trophoblasts increased infection resistance. Our study has significant implications for understanding the etiology of placental infections and prematurity and developing strategies to mitigate placental infection-induced PTB.
Conflict of interest statement
The authors have declared that no conflict of interest exists.
Figures


Similar articles
-
Impaired autophagy with augmented apoptosis in a Th1/Th2-imbalanced placental micromilieu is associated with spontaneous preterm birth.Front Mol Biosci. 2022 Aug 26;9:897228. doi: 10.3389/fmolb.2022.897228. eCollection 2022. Front Mol Biosci. 2022. PMID: 36090032 Free PMC article.
-
Autophagy as a mechanism of antiviral defense at the maternal-fetal interface.Autophagy. 2013 Dec;9(12):2173-4. doi: 10.4161/auto.26558. Epub 2013 Oct 8. Autophagy. 2013. PMID: 24231730
-
The placental factor in spontaneous preterm birth in twin vs. singleton pregnancies.Eur J Obstet Gynecol Reprod Biol. 2017 Jul;214:1-5. doi: 10.1016/j.ejogrb.2017.04.035. Epub 2017 Apr 22. Eur J Obstet Gynecol Reprod Biol. 2017. PMID: 28448801
-
Gestational food restriction decreases placental interleukin-10 expression and markers of autophagy and endoplasmic reticulum stress in murine intrauterine growth restriction.Nutr Res. 2016 Oct;36(10):1055-1067. doi: 10.1016/j.nutres.2016.08.001. Epub 2016 Aug 5. Nutr Res. 2016. PMID: 27865347 Free PMC article.
-
Autophagy regulation of physiological and pathological processes in the female reproductive tract.Am J Reprod Immunol. 2017 May;77(5). doi: 10.1111/aji.12650. Epub 2017 Feb 13. Am J Reprod Immunol. 2017. PMID: 28194822 Review.
Cited by
-
Homeostasis Maintenance in Plasmodium-Infected Placentas: Is There a Role for Placental Autophagy During Malaria in Pregnancy?Front Immunol. 2022 Jul 11;13:931034. doi: 10.3389/fimmu.2022.931034. eCollection 2022. Front Immunol. 2022. PMID: 35898514 Free PMC article.
-
Zika virus NS1 drives tunneling nanotube formation for mitochondrial transfer and stealth transmission in trophoblasts.Nat Commun. 2025 Feb 20;16(1):1803. doi: 10.1038/s41467-025-56927-2. Nat Commun. 2025. PMID: 39979240 Free PMC article.
-
Hypoxia-Reoxygenation Impairs Autophagy-Lysosomal Machinery in Primary Human Trophoblasts Mimicking Placental Pathology of Early-Onset Preeclampsia.Int J Mol Sci. 2022 May 18;23(10):5644. doi: 10.3390/ijms23105644. Int J Mol Sci. 2022. PMID: 35628454 Free PMC article.
-
Stage-specific autophagy dynamics in reproductive processes and associated disorders.Front Cell Dev Biol. 2025 Jul 28;13:1639691. doi: 10.3389/fcell.2025.1639691. eCollection 2025. Front Cell Dev Biol. 2025. PMID: 40791986 Free PMC article. Review.
-
Host and viral mechanisms of congenital Zika syndrome.Virulence. 2019 Dec;10(1):768-775. doi: 10.1080/21505594.2019.1656503. Virulence. 2019. PMID: 31451049 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
