

Robust and scalable inference of population history from hundreds of unphased whole genomes
- PMID: 28024154
- PMCID: PMC5470542
- DOI: 10.1038/ng.3748
Robust and scalable inference of population history from hundreds of unphased whole genomes
Abstract
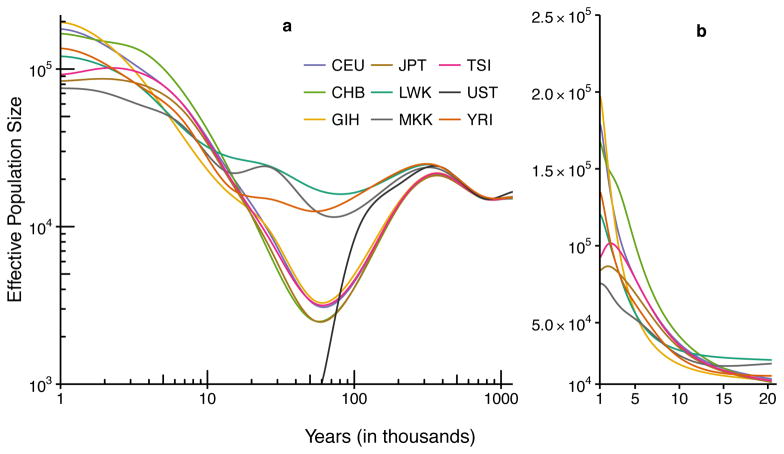
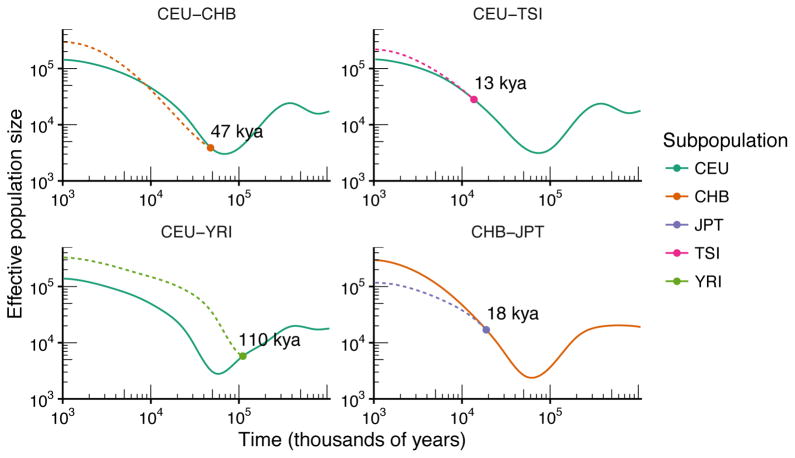
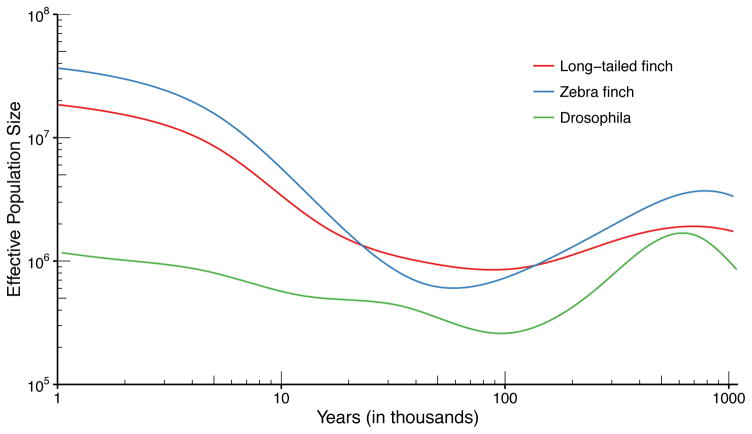
It has recently been demonstrated that inference methods based on genealogical processes with recombination can uncover past population history in unprecedented detail. However, these methods scale poorly with sample size, limiting resolution in the recent past, and they require phased genomes, which contain switch errors that can catastrophically distort the inferred history. Here we present SMC++, a new statistical tool capable of analyzing orders of magnitude more samples than existing methods while requiring only unphased genomes (its results are independent of phasing). SMC++ can jointly infer population size histories and split times in diverged populations, and it employs a novel spline regularization scheme that greatly reduces estimation error. We apply SMC++ to analyze sequence data from over a thousand human genomes in Africa and Eurasia, hundreds of genomes from a Drosophila melanogaster population in Africa, and tens of genomes from zebra finch and long-tailed finch populations in Australia.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
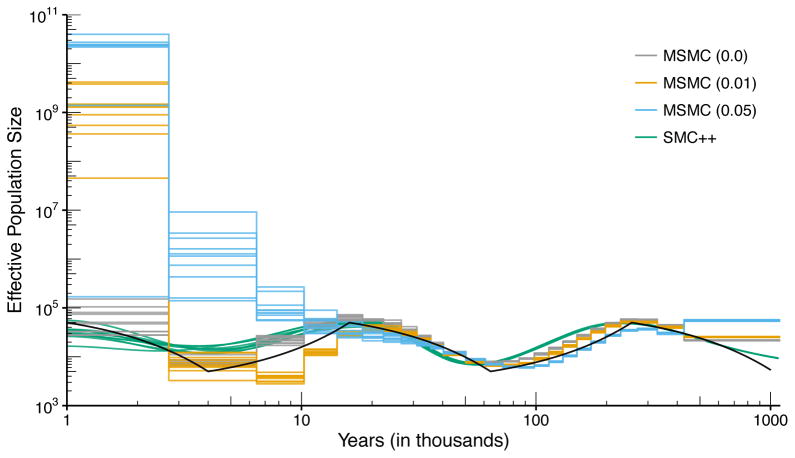
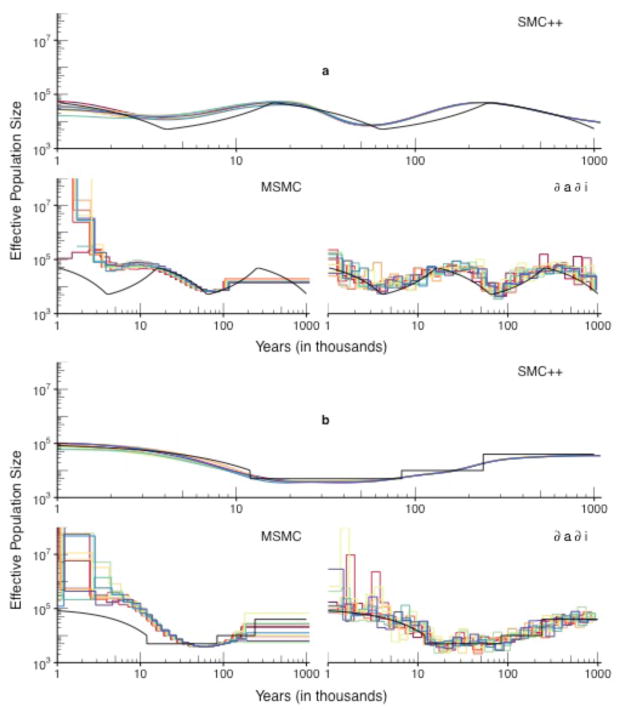
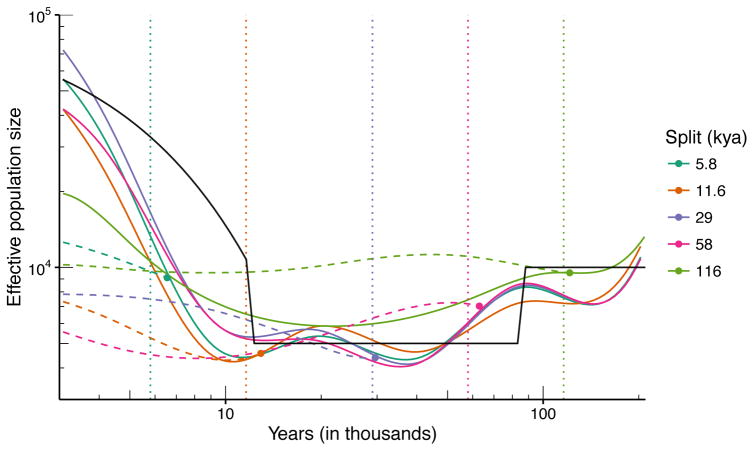
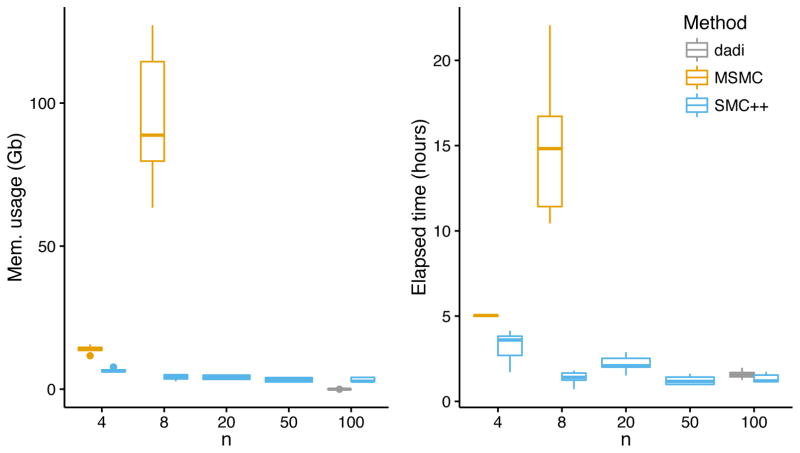
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
