

Variable chromatin structure revealed by in situ spatially correlated DNA cleavage mapping
- PMID: 28024297
- PMCID: PMC5526328
- DOI: 10.1038/nature20781
Variable chromatin structure revealed by in situ spatially correlated DNA cleavage mapping
Abstract
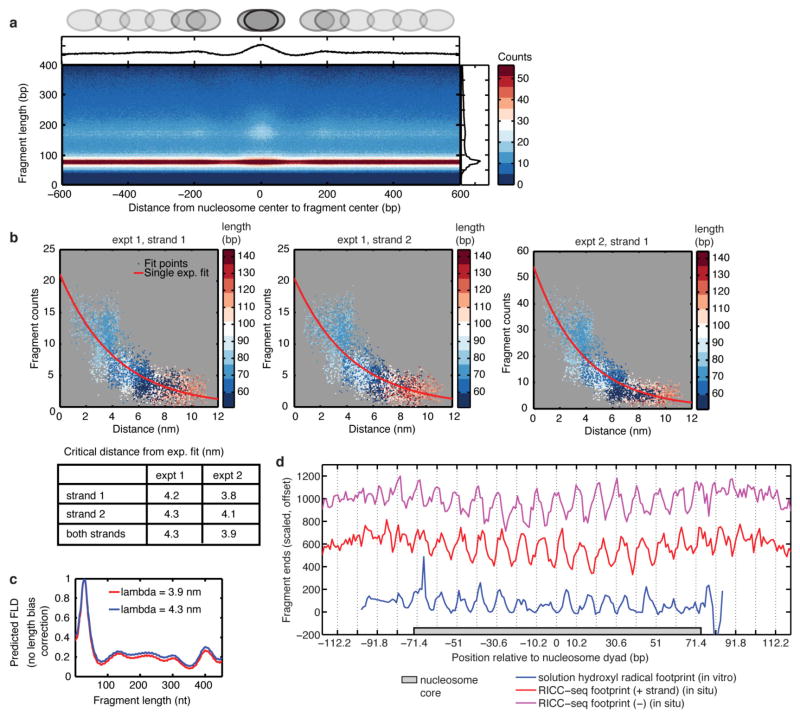
Chromatin structure at the length scale encompassing local nucleosome-nucleosome interactions is thought to play a crucial role in regulating transcription and access to DNA. However, this secondary structure of chromatin remains poorly understood compared with the primary structure of single nucleosomes or the tertiary structure of long-range looping interactions. Here we report the first genome-wide map of chromatin conformation in human cells at the 1-3 nucleosome (50-500 bp) scale, obtained using ionizing radiation-induced spatially correlated cleavage of DNA with sequencing (RICC-seq) to identify DNA-DNA contacts that are spatially proximal. Unbiased analysis of RICC-seq signal reveals regional enrichment of DNA fragments characteristic of alternating rather than adjacent nucleosome interactions in tri-nucleosome units, particularly in H3K9me3-marked heterochromatin. We infer differences in the likelihood of nucleosome-nucleosome contacts among open chromatin, H3K27me3-marked, and H3K9me3-marked repressed chromatin regions. After calibrating RICC-seq signal to three-dimensional distances, we show that compact two-start helical fibre structures with stacked alternating nucleosomes are consistent with RICC-seq fragmentation patterns from H3K9me3-marked chromatin, while non-compact structures and solenoid structures are consistent with open chromatin. Our data support a model of chromatin architecture in intact interphase nuclei consistent with variable longitudinal compaction of two-start helical fibres.
Conflict of interest statement
The authors declare competing financial interests: details are available in the online version of the paper.
Figures













References
-
- Gilbert N, et al. Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell. 2004;118:555–566. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
