

Population Parameters Underlying an Ongoing Soft Sweep in Southeast Asian Malaria Parasites
- PMID: 28025270
- PMCID: PMC5216669
- DOI: 10.1093/molbev/msw228
Population Parameters Underlying an Ongoing Soft Sweep in Southeast Asian Malaria Parasites
Abstract
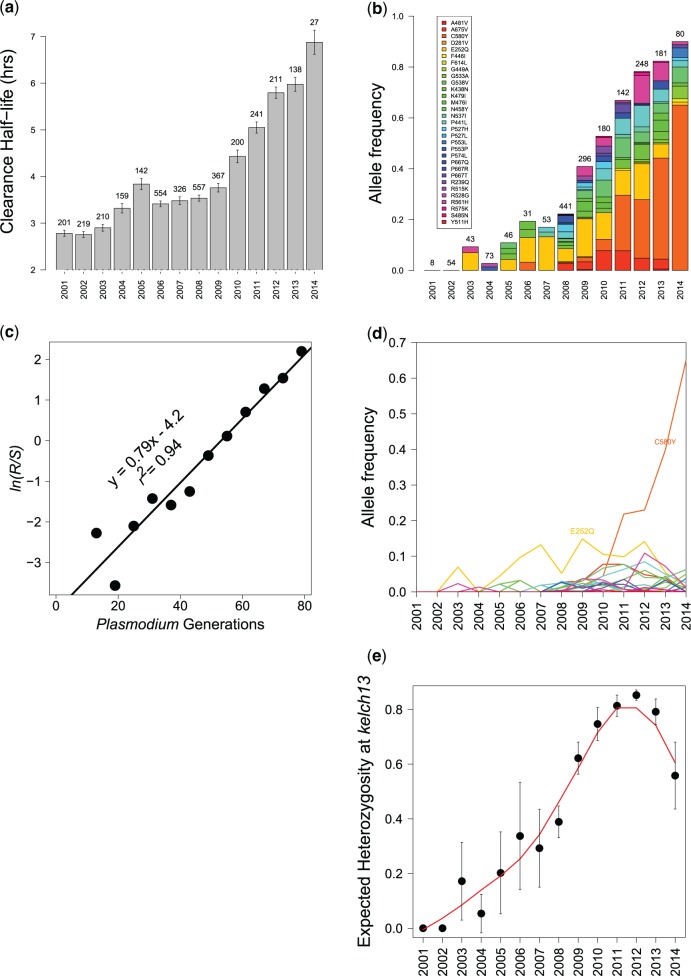
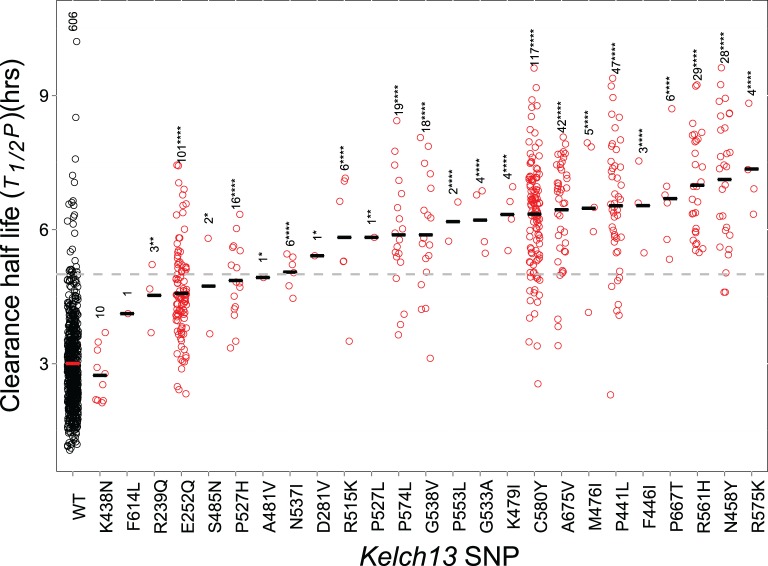
Multiple kelch13 alleles conferring artemisinin resistance (ART-R) are currently spreading through Southeast Asian malaria parasite populations, providing a unique opportunity to observe an ongoing soft selective sweep, investigate why resistance alleles have evolved multiple times and determine fundamental population genetic parameters for Plasmodium We sequenced kelch13 (n = 1,876), genotyped 75 flanking SNPs, and measured clearance rate (n = 3,552) in parasite infections from Western Thailand (2001-2014). We describe 32 independent coding mutations including common mutations outside the kelch13 propeller associated with significant reductions in clearance rate. Mutations were first observed in 2003 and rose to 90% by 2014, consistent with a selection coefficient of ∼0.079. ART-R allele diversity rose until 2012 and then dropped as one allele (C580Y) spread to high frequency. The frequency with which adaptive alleles arise is determined by the rate of mutation and the population size. Two factors drive this soft sweep: (1) multiple kelch13 amino-acid mutations confer resistance providing a large mutational target-we estimate the target is 87-163 bp. (2) The population mutation parameter (Θ = 2Neμ) can be estimated from the frequency distribution of ART-R alleles and is ∼5.69, suggesting that short term effective population size is 88 thousand to 1.2 million. This is 52-705 times greater than Ne estimated from fluctuation in allele frequencies, suggesting that we have previously underestimated the capacity for adaptive evolution in Plasmodium Our central conclusions are that retrospective studies may underestimate the complexity of selective events and the Ne relevant for adaptation for malaria is considerably higher than previously estimated.
Keywords: adaptation; drug resistance; effective population size; plasmodium; rapid evolution; soft selective sweep.
© The Author 2016. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures



References
-
- Anderson TJ, Roper C. 2005. The origins and spread of antimalarial drug resistance: lessons for policy makers. Acta Trop. 94:269–280. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
