

Emerging therapies and challenges in spinal muscular atrophy
- PMID: 28026041
- PMCID: PMC5396275
- DOI: 10.1002/ana.24864
Emerging therapies and challenges in spinal muscular atrophy
Abstract
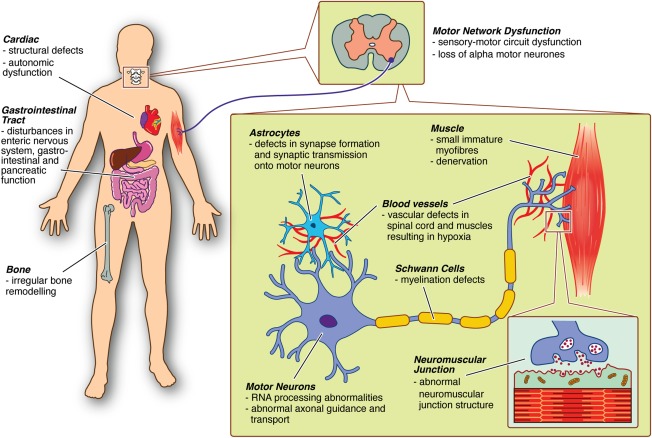
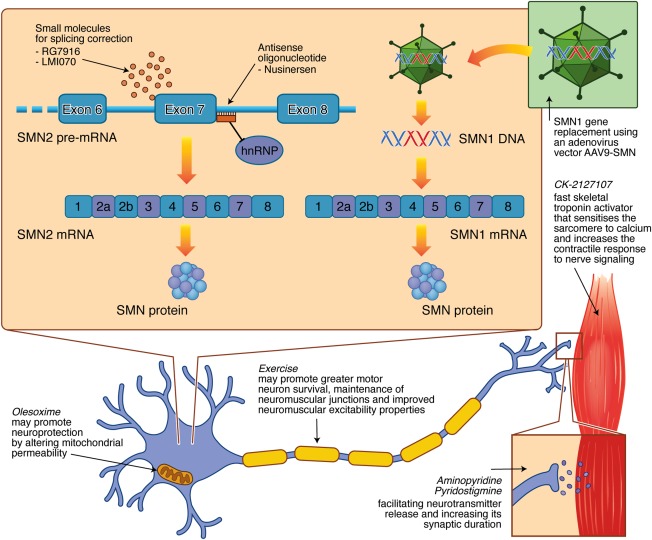
Spinal muscular atrophy (SMA) is a hereditary neurodegenerative disease with severity ranging from progressive infantile paralysis and premature death (type I) to limited motor neuron loss and normal life expectancy (type IV). Without disease-modifying therapies, the impact is profound for patients and their families. Improved understanding of the molecular basis of SMA, disease pathogenesis, natural history, and recognition of the impact of standardized care on outcomes has yielded progress toward the development of novel therapeutic strategies and are summarized. Therapeutic strategies in the pipeline are appraised, ranging from SMN1 gene replacement to modulation of SMN2 encoded transcripts, to neuroprotection, to an expanding repertoire of peripheral targets, including muscle. With the advent of preliminary trial data, it can be reasonably anticipated that the SMA treatment landscape will transform significantly. Advancement in presymptomatic diagnosis and screening programs will be critical, with pilot newborn screening studies underway to facilitate preclinical diagnosis. The development of disease-modifying therapies will necessitate monitoring programs to determine the long-term impact, careful evaluation of combined treatments, and further acceleration of improvements in supportive care. In advance of upcoming clinical trial results, we consider the challenges and controversies related to the implementation of novel therapies for all patients and set the scene as the field prepares to enter an era of novel therapies. Ann Neurol 2017;81:355-368.
© 2016 The Authors. Annals of Neurology published by Wiley Periodicals, Inc. on behalf of American Neurological Association.
Figures
References
-
- Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy‐determining gene. Cell 1995;80:155–165. - PubMed
-
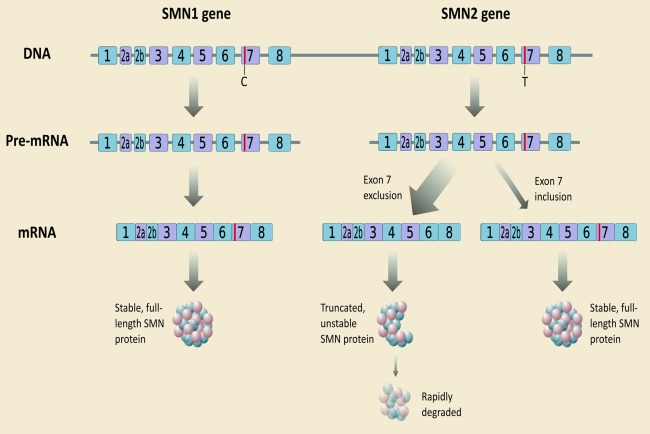
- Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 1999;8:1177–1183. - PubMed
-
- Dunaway S, Montes J, Ryan PA, et al. Spinal muscular atrophy type III: trying to understand subtle functional change over time: a case report. J Child Neurol 2012;27:779–785. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
