

Somatic Mutation Patterns in Hemizygous Genomic Regions Unveil Purifying Selection during Tumor Evolution
- PMID: 28027311
- PMCID: PMC5189942
- DOI: 10.1371/journal.pgen.1006506
Somatic Mutation Patterns in Hemizygous Genomic Regions Unveil Purifying Selection during Tumor Evolution
Abstract
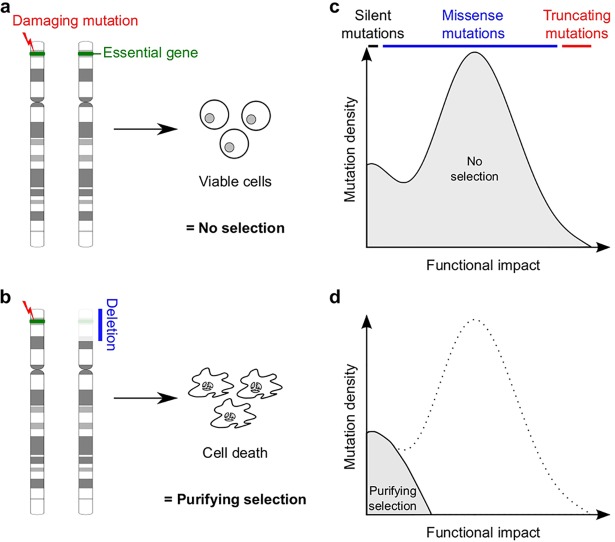
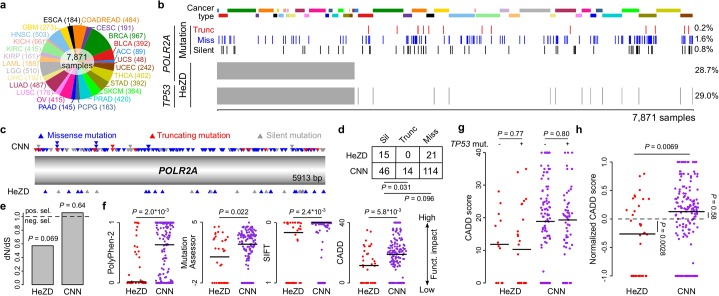
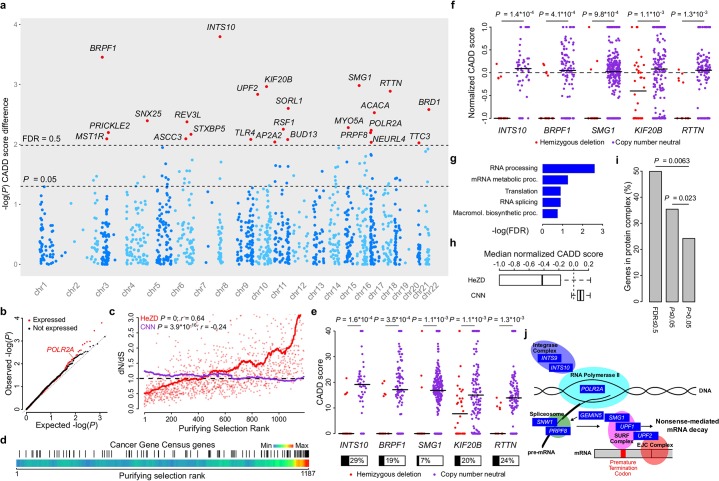
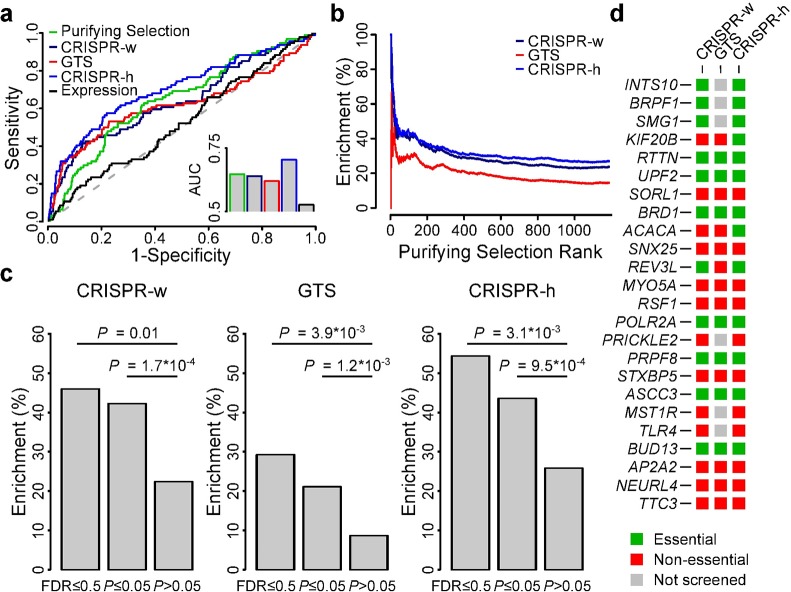
Identification of cancer driver genes using somatic mutation patterns indicative of positive selection has become a major goal in cancer genomics. However, cancer cells additionally depend on a large number of genes involved in basic cellular processes. While such genes should in theory be subject to strong purifying (negative) selection against damaging somatic mutations, these patterns have been elusive and purifying selection remains inadequately explored in cancer. Here, we hypothesized that purifying selection should be evident in hemizygous genomic regions, where damaging mutations cannot be compensated for by healthy alleles. Using a 7,781-sample pan-cancer dataset, we first confirmed this in POLR2A, an essential gene where hemizygous deletions are known to confer elevated sensitivity to pharmacological suppression. We next used this principle to identify several genes and pathways that show patterns indicative of purifying selection to avoid deleterious mutations. These include the POLR2A interacting protein INTS10 as well as genes involved in mRNA splicing, nonsense-mediated mRNA decay and other RNA processing pathways. Many of these genes belong to large protein complexes, and strong overlaps were observed with recent functional screens for gene essentiality in human cells. Our analysis supports that purifying selection acts to preserve the remaining function of many hemizygously deleted essential genes in tumors, indicating vulnerabilities that might be exploited by future therapeutic strategies.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
