

Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms
- PMID: 28028026
- PMCID: PMC5335805
- DOI: 10.1182/blood-2016-10-695957
Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms
Abstract
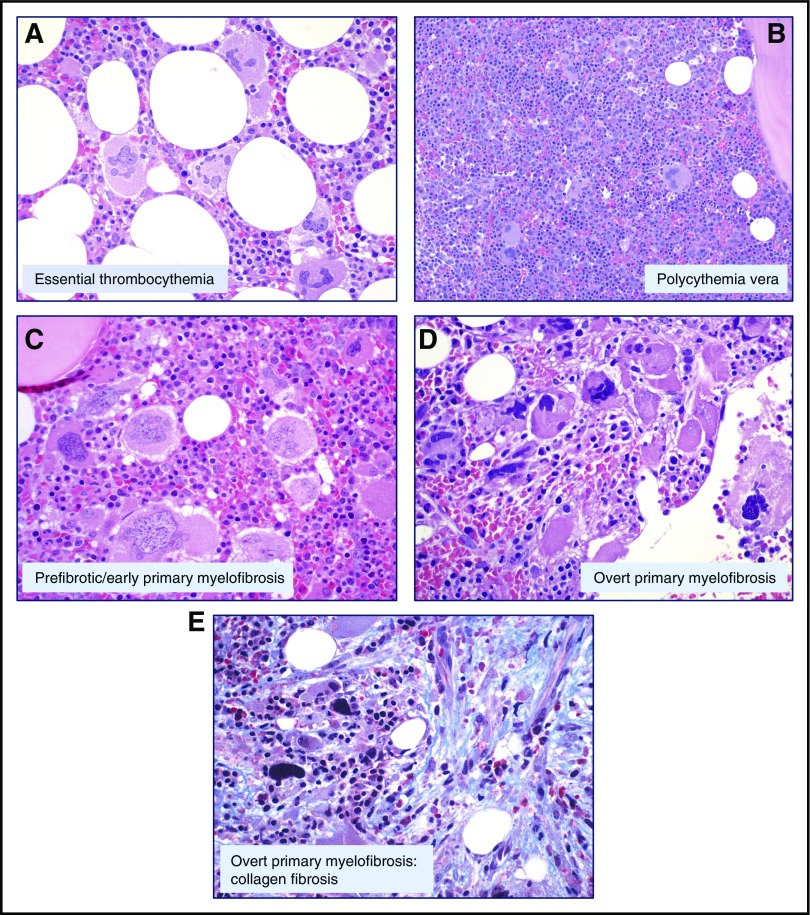
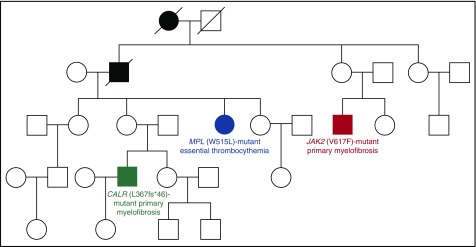
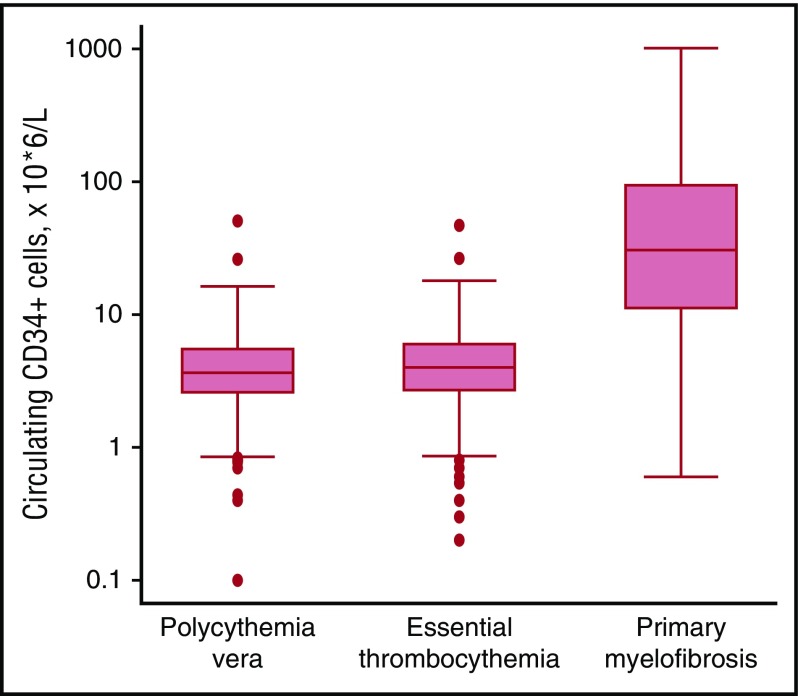
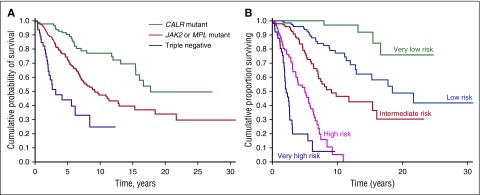
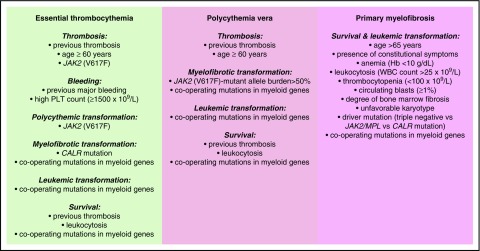
Philadelphia-negative classical myeloproliferative neoplasms (MPNs) include polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). The 2016 revision of the WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues includes new criteria for the diagnosis of these disorders. Somatic mutations in the 3 driver genes, that is, JAK2, CALR, and MPL, represent major diagnostic criteria in combination with hematologic and morphological abnormalities. PV is characterized by erythrocytosis with suppressed endogenous erythropoietin production, bone marrow panmyelosis, and JAK2 mutation. Thrombocytosis, bone marrow megakaryocytic proliferation, and presence of JAK2, CALR, or MPL mutation are the main diagnostic criteria for ET. PMF is characterized by bone marrow megakaryocytic proliferation, reticulin and/or collagen fibrosis, and presence of JAK2, CALR, or MPL mutation. Prefibrotic myelofibrosis represents an early phase of myelofibrosis, and is characterized by granulocytic/megakaryocytic proliferation and lack of reticulin fibrosis in the bone marrow. The genomic landscape of MPNs is more complex than initially thought and involves several mutant genes beyond the 3 drivers. Comutated, myeloid tumor-suppressor genes contribute to phenotypic variability, phenotypic shifts, and progression to more aggressive disorders. Patients with myeloid neoplasms are at variable risk of vascular complications, including arterial or venous thrombosis and bleeding. Current prognostic models are mainly based on clinical and hematologic parameters, but innovative models that include genetic data are being developed for both clinical and trial settings. In perspective, molecular profiling of MPNs might also allow for accurate evaluation and monitoring of response to innovative drugs that target the mutant clone.
© 2017 by The American Society of Hematology.
Figures
References
-
- Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951;6(4):372-375. - PubMed
-
- Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC; 2008.
-
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. - PubMed
-
- James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144-1148. - PubMed
-
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779-1790. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
