

Evolution-informed modeling improves outcome prediction for cancers
- PMID: 28035236
- PMCID: PMC5192825
- DOI: 10.1111/eva.12417
Evolution-informed modeling improves outcome prediction for cancers
Abstract
Despite wide applications of high-throughput biotechnologies in cancer research, many biomarkers discovered by exploring large-scale omics data do not provide satisfactory performance when used to predict cancer treatment outcomes. This problem is partly due to the overlooking of functional implications of molecular markers. Here, we present a novel computational method that uses evolutionary conservation as prior knowledge to discover bona fide biomarkers. Evolutionary selection at the molecular level is nature's test on functional consequences of genetic elements. By prioritizing genes that show significant statistical association and high functional impact, our new method reduces the chances of including spurious markers in the predictive model. When applied to predicting therapeutic responses for patients with acute myeloid leukemia and to predicting metastasis for patients with prostate cancers, the new method gave rise to evolution-informed models that enjoyed low complexity and high accuracy. The identified genetic markers also have significant implications in tumor progression and embrace potential drug targets. Because evolutionary conservation can be estimated as a gene-specific, position-specific, or allele-specific parameter on the nucleotide level and on the protein level, this new method can be extended to apply to miscellaneous "omics" data to accelerate biomarker discoveries.
Keywords: evolutionary medicine; genomics/proteomics; molecular evolution; transcriptomics.
Figures
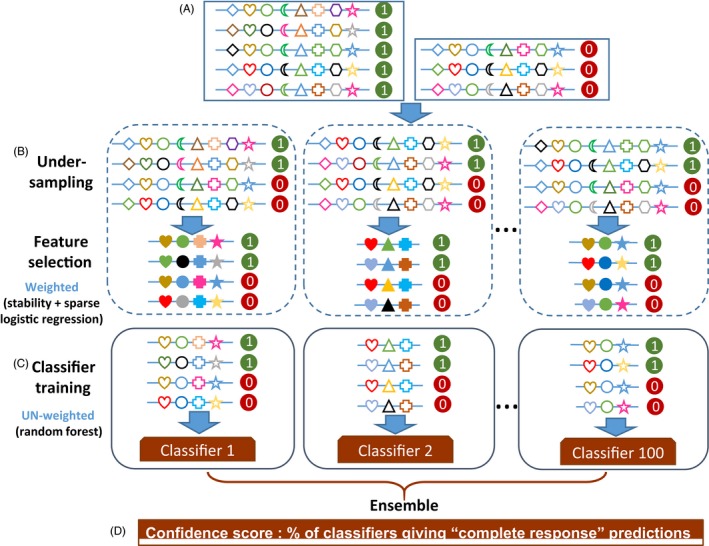
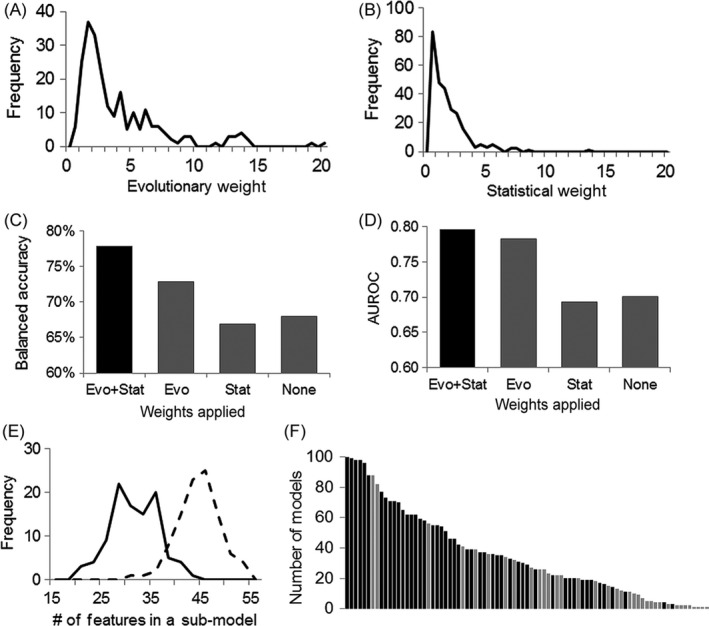
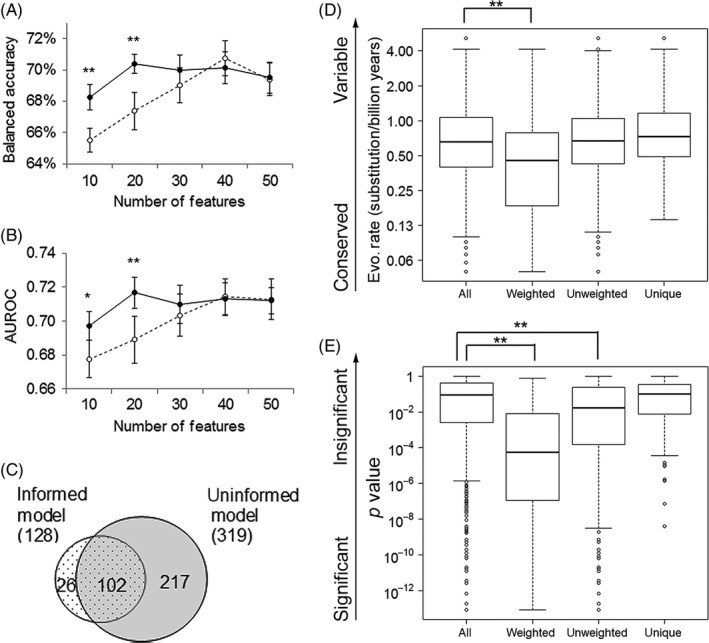
References
-
- Banerji, V. , Frumm, S. M. , Ross, K. N. , Li, L. S. , Schinzel, A. C. , Hahn, C. K. , … Stegmaier, K . (2012). The intersection of genetic and chemical genomic screens identifies GSK‐3alpha as a target in human acute myeloid leukemia. The Journal of Clinical Investigation, 122, 935–947. - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
