

Heparanase augments insulin receptor signaling in breast carcinoma
- PMID: 28038446
- PMCID: PMC5386693
- DOI: 10.18632/oncotarget.14292
Heparanase augments insulin receptor signaling in breast carcinoma
Abstract
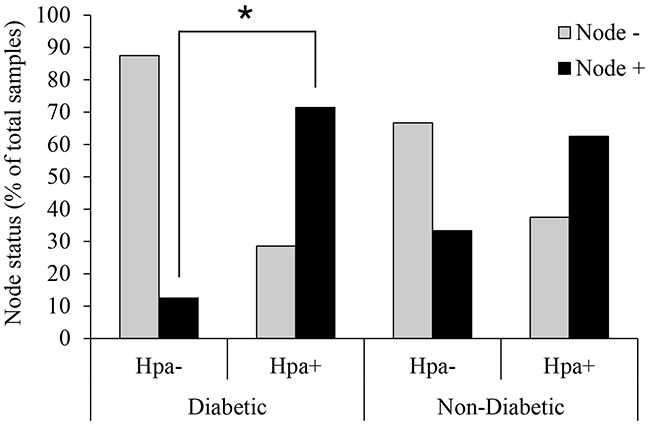
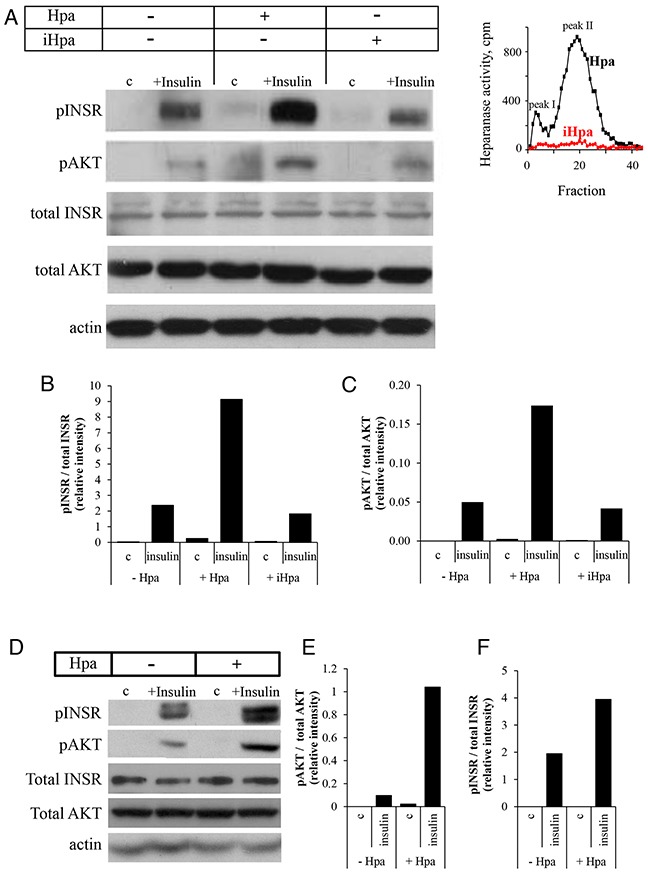
Recently, growing interest in the potential link between metabolic disorders (i.e., diabetes, obesity, metabolic syndrome) and breast cancer has mounted, including studies which indicate that diabetic/hyperinsulinemic women have a significantly higher risk of bearing breast tumors that are more aggressive and associated with higher death rates. Insulin signaling is regarded as a major contributor to this phenomenon; much less is known about the role of heparan sulfate-degrading enzyme heparanase in the link between metabolic disorders and cancer.In the present study we analyzed clinical samples of breast carcinoma derived from diabetic/non-diabetic patients, and investigated effects of heparanase on insulin signaling in breast carcinoma cell lines, as well as insulin-driven growth of breast tumor cells.We demonstrate that heparanase activity leads to enhanced insulin signaling and activation of downstream tumor-promoting pathways in breast carcinoma cells. In agreement, heparanase enhances insulin-induced proliferation of breast tumor cells in vitro. Moreover, analyzing clinical data from diabetic breast carcinoma patients, we found that concurrent presence of both diabetic state and heparanase in tumor tissue (as opposed to either condition alone) was associated with more aggressive phenotype of breast tumors in the patient cohort analyzed in our study (two-sided Fisher's exact test; p=0.04). Our findings highlight the emerging role of heparanase in powering effect of hyperinsulinemic state on breast tumorigenesis and imply that heparanase targeting, which is now under intensive development/clinical testing, could be particularly efficient in a growing fraction of breast carcinoma patients suffering from metabolic disorders.
Keywords: breast carcinoma; diabetes; heparanase; insulin receptor.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

References
-
- Hursting SD. Obesity, energy balance, and cancer: a mechanistic perspective. Cancer Treat Res. 2014;159:21–33. - PubMed
-
- Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nat Rev Cancer. 2011;11:886–95. - PubMed
-
- Lipscombe L, Fischer H, Austin P, Fu L, Jaakkimainen RL, Ginsburg O, Rochon P, Narod S, Paszat L. The association between diabetes and breast cancer stage at diagnosis: a population-based study. Breast Cancer Research and Treatment. 2015;150:613–20. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
