

An Unbiased Estimator of Gene Diversity with Improved Variance for Samples Containing Related and Inbred Individuals of any Ploidy
- PMID: 28040781
- PMCID: PMC5295611
- DOI: 10.1534/g3.116.037168
An Unbiased Estimator of Gene Diversity with Improved Variance for Samples Containing Related and Inbred Individuals of any Ploidy
Abstract
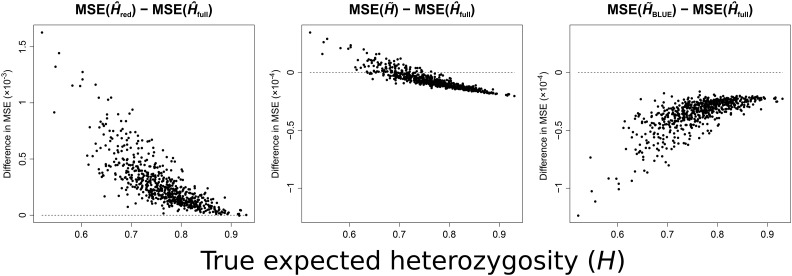
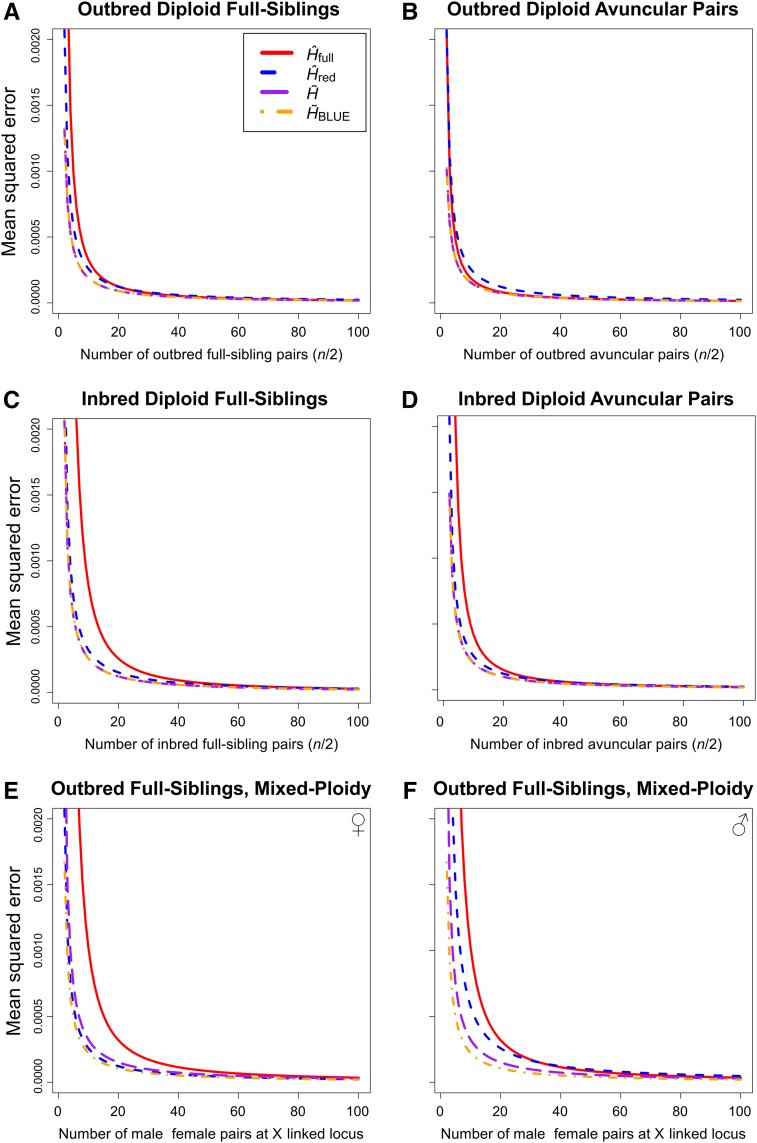
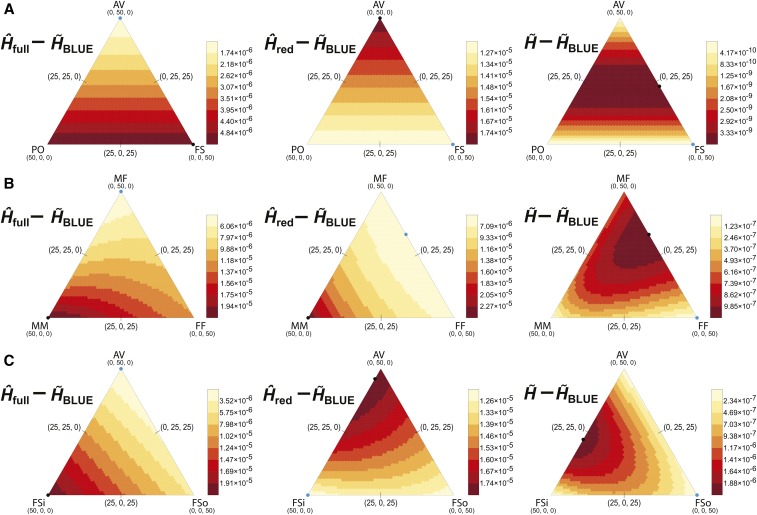
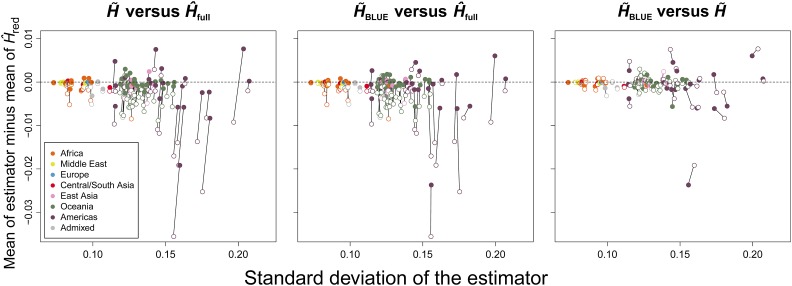
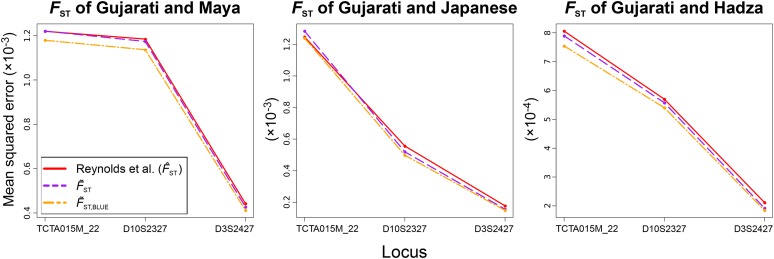
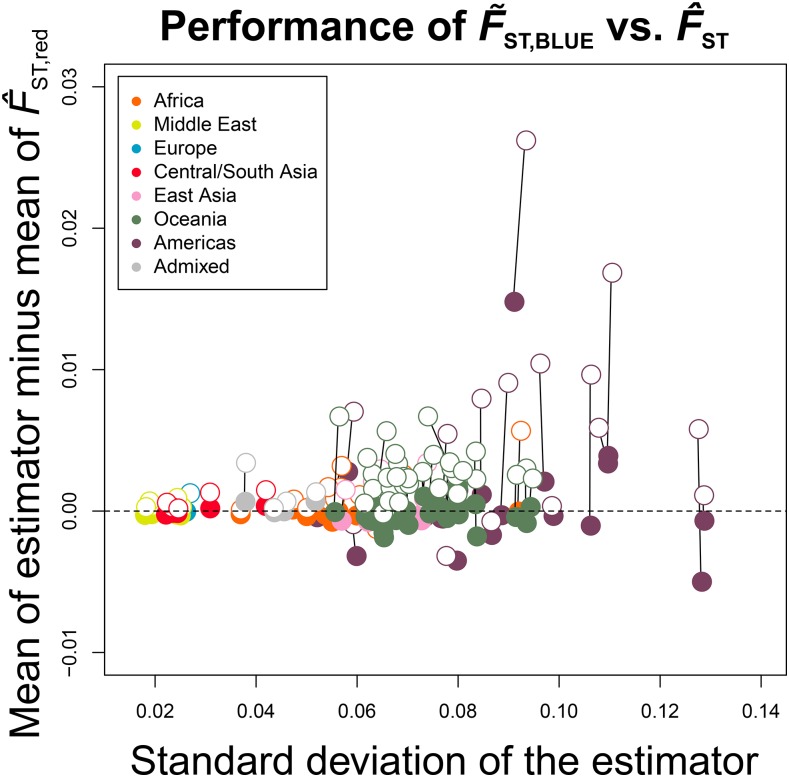
Gene diversity, or expected heterozygosity (H), is a common statistic for assessing genetic variation within populations. Estimation of this statistic decreases in accuracy and precision when individuals are related or inbred, due to increased dependence among allele copies in the sample. The original unbiased estimator of expected heterozygosity underestimates true population diversity in samples containing relatives, as it only accounts for sample size. More recently, a general unbiased estimator of expected heterozygosity was developed that explicitly accounts for related and inbred individuals in samples. Though unbiased, this estimator's variance is greater than that of the original estimator. To address this issue, we introduce a general unbiased estimator of gene diversity for samples containing related or inbred individuals, which employs the best linear unbiased estimator of allele frequencies, rather than the commonly used sample proportion. We examine the properties of this estimator, [Formula: see text] relative to alternative estimators using simulations and theoretical predictions, and show that it predominantly has the smallest mean squared error relative to others. Further, we empirically assess the performance of [Formula: see text] on a global human microsatellite dataset of 5795 individuals, from 267 populations, genotyped at 645 loci. Additionally, we show that the improved variance of [Formula: see text] leads to improved estimates of the population differentiation statistic, [Formula: see text] which employs measures of gene diversity within its calculation. Finally, we provide an R script, BestHet, to compute this estimator from genomic and pedigree data.
Keywords: expected heterozygosity; identity state; inbreeding; locus-specific branch length; relatedness.
Copyright © 2017 Harris and DeGiorgio.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources