

Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease
- PMID: 28041643
- PMCID: PMC5223092
- DOI: 10.1016/j.ajhg.2016.12.003
Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease
Abstract
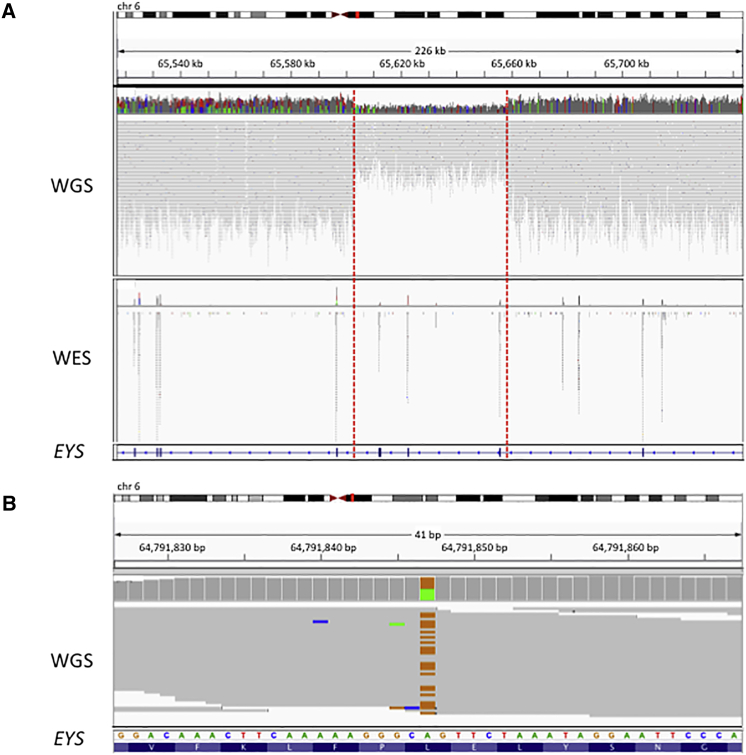
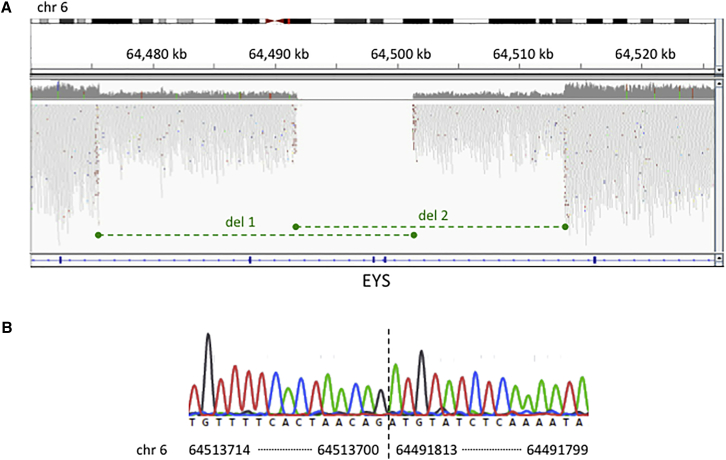
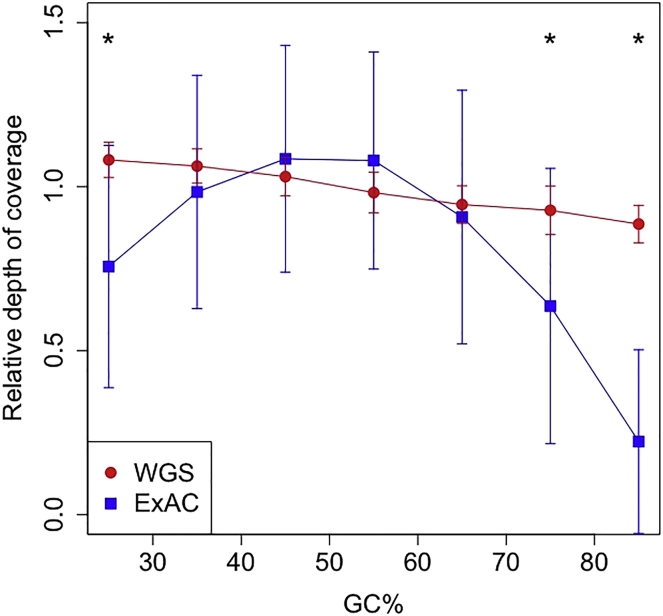
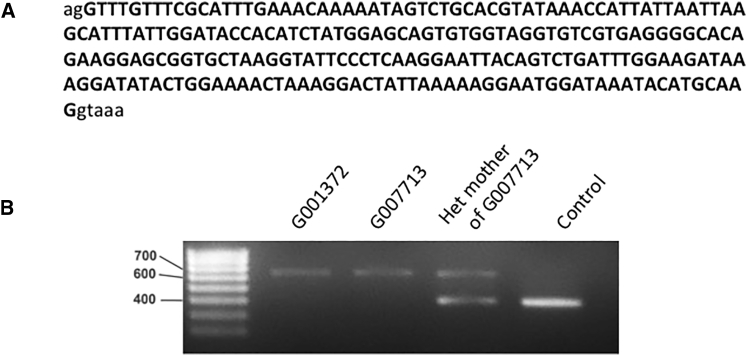
Inherited retinal disease is a common cause of visual impairment and represents a highly heterogeneous group of conditions. Here, we present findings from a cohort of 722 individuals with inherited retinal disease, who have had whole-genome sequencing (n = 605), whole-exome sequencing (n = 72), or both (n = 45) performed, as part of the NIHR-BioResource Rare Diseases research study. We identified pathogenic variants (single-nucleotide variants, indels, or structural variants) for 404/722 (56%) individuals. Whole-genome sequencing gives unprecedented power to detect three categories of pathogenic variants in particular: structural variants, variants in GC-rich regions, which have significantly improved coverage compared to whole-exome sequencing, and variants in non-coding regulatory regions. In addition to previously reported pathogenic regulatory variants, we have identified a previously unreported pathogenic intronic variant in CHM in two males with choroideremia. We have also identified 19 genes not previously known to be associated with inherited retinal disease, which harbor biallelic predicted protein-truncating variants in unsolved cases. Whole-genome sequencing is an increasingly important comprehensive method with which to investigate the genetic causes of inherited retinal disease.
Keywords: copy-number variants; rare sequence variant; retinal dystrophy; whole-genome sequence.
Copyright © 2017. Published by Elsevier Inc.
Figures


References
-
- Beaulieu C.L., Majewski J., Schwartzentruber J., Samuels M.E., Fernandez B.A., Bernier F.P., Brudno M., Knoppers B., Marcadier J., Dyment D., FORGE Canada Consortium FORGE Canada Consortium: outcomes of a 2-year national rare-disease gene-discovery project. Am. J. Hum. Genet. 2014;94:809–817. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
