

ChimPipe: accurate detection of fusion genes and transcription-induced chimeras from RNA-seq data
- PMID: 28049418
- PMCID: PMC5209911
- DOI: 10.1186/s12864-016-3404-9
ChimPipe: accurate detection of fusion genes and transcription-induced chimeras from RNA-seq data
Abstract
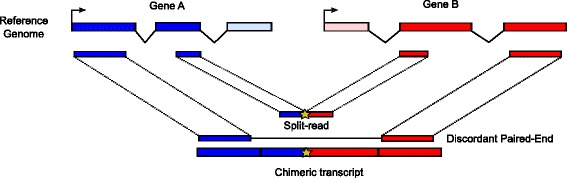
Background: Chimeric transcripts are commonly defined as transcripts linking two or more different genes in the genome, and can be explained by various biological mechanisms such as genomic rearrangement, read-through or trans-splicing, but also by technical or biological artefacts. Several studies have shown their importance in cancer, cell pluripotency and motility. Many programs have recently been developed to identify chimeras from Illumina RNA-seq data (mostly fusion genes in cancer). However outputs of different programs on the same dataset can be widely inconsistent, and tend to include many false positives. Other issues relate to simulated datasets restricted to fusion genes, real datasets with limited numbers of validated cases, result inconsistencies between simulated and real datasets, and gene rather than junction level assessment.
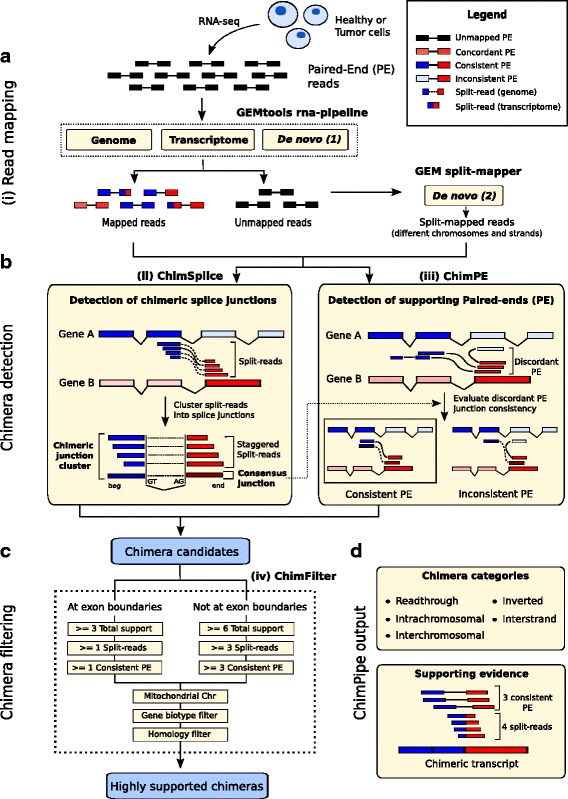
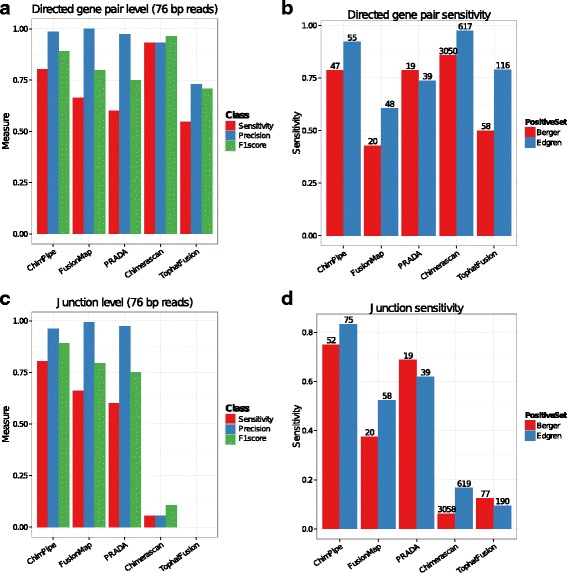
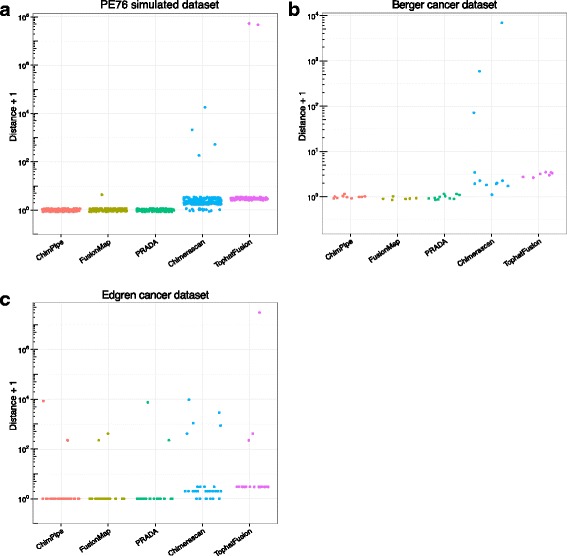
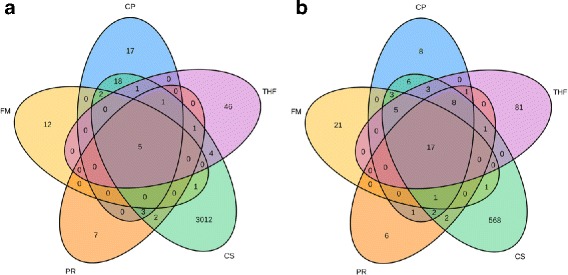
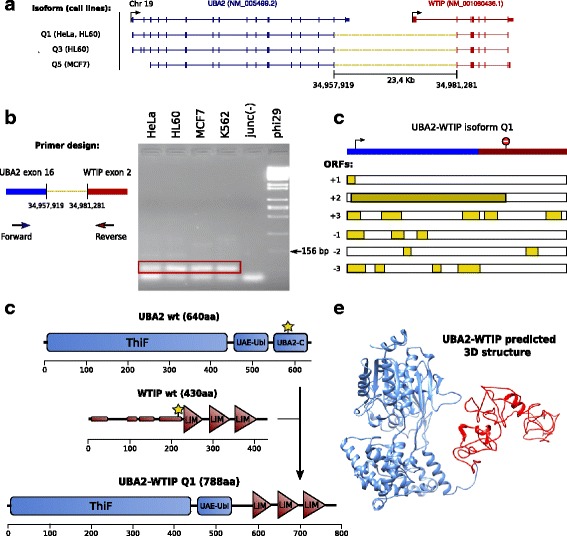
Results: Here we present ChimPipe, a modular and easy-to-use method to reliably identify fusion genes and transcription-induced chimeras from paired-end Illumina RNA-seq data. We have also produced realistic simulated datasets for three different read lengths, and enhanced two gold-standard cancer datasets by associating exact junction points to validated gene fusions. Benchmarking ChimPipe together with four other state-of-the-art tools on this data showed ChimPipe to be the top program at identifying exact junction coordinates for both kinds of datasets, and the one showing the best trade-off between sensitivity and precision. Applied to 106 ENCODE human RNA-seq datasets, ChimPipe identified 137 high confidence chimeras connecting the protein coding sequence of their parent genes. In subsequent experiments, three out of four predicted chimeras, two of which recurrently expressed in a large majority of the samples, could be validated. Cloning and sequencing of the three cases revealed several new chimeric transcript structures, 3 of which with the potential to encode a chimeric protein for which we hypothesized a new role. Applying ChimPipe to human and mouse ENCODE RNA-seq data led to the identification of 131 recurrent chimeras common to both species, and therefore potentially conserved.
Conclusions: ChimPipe combines discordant paired-end reads and split-reads to detect any kind of chimeras, including those originating from polymerase read-through, and shows an excellent trade-off between sensitivity and precision. The chimeras found by ChimPipe can be validated in-vitro with high accuracy.
Keywords: Benchmark; Cancer; Chimera; Fusion gene; Isoform; RNA-seq; Simulation; Splice junction; Transcript.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
