

Hereditary Lysozyme Amyloidosis Variant p.Leu102Ser Associates with Unique Phenotype
- PMID: 28049649
- PMCID: PMC5280032
- DOI: 10.1681/ASN.2016090951
Hereditary Lysozyme Amyloidosis Variant p.Leu102Ser Associates with Unique Phenotype
Abstract
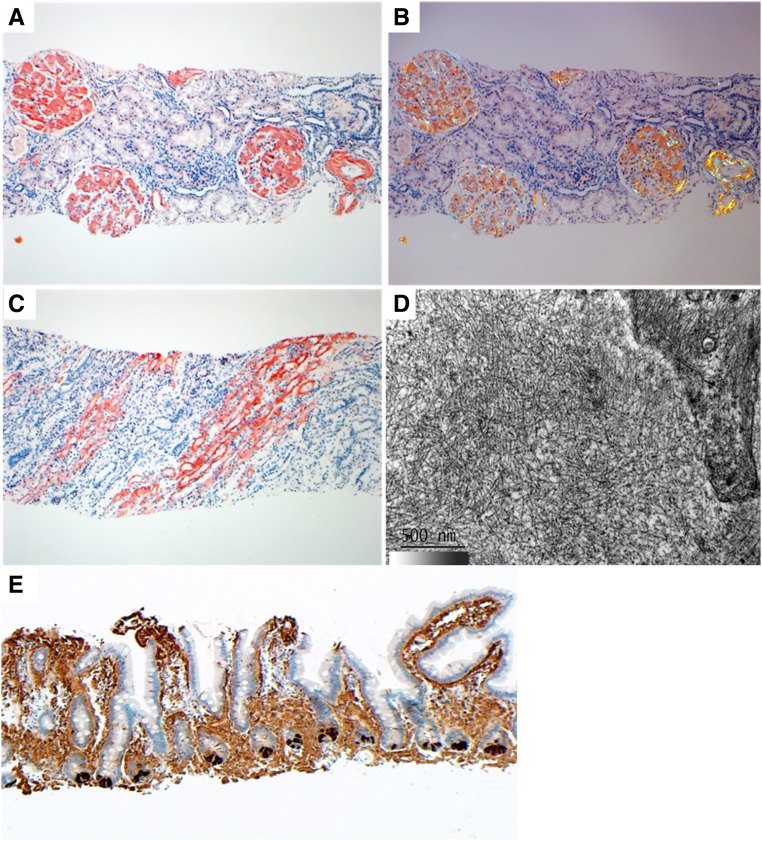
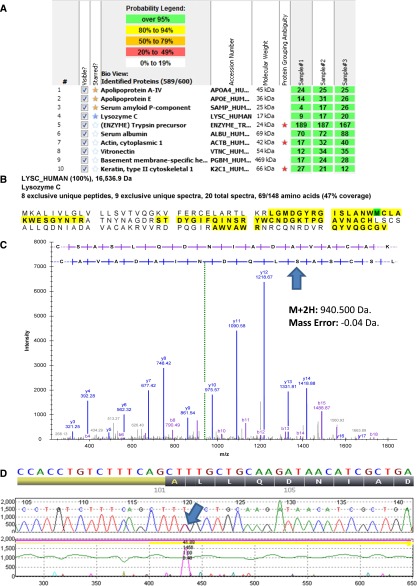
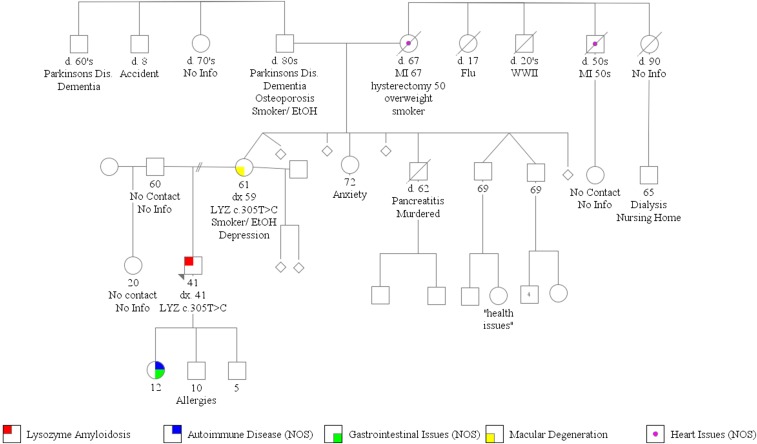
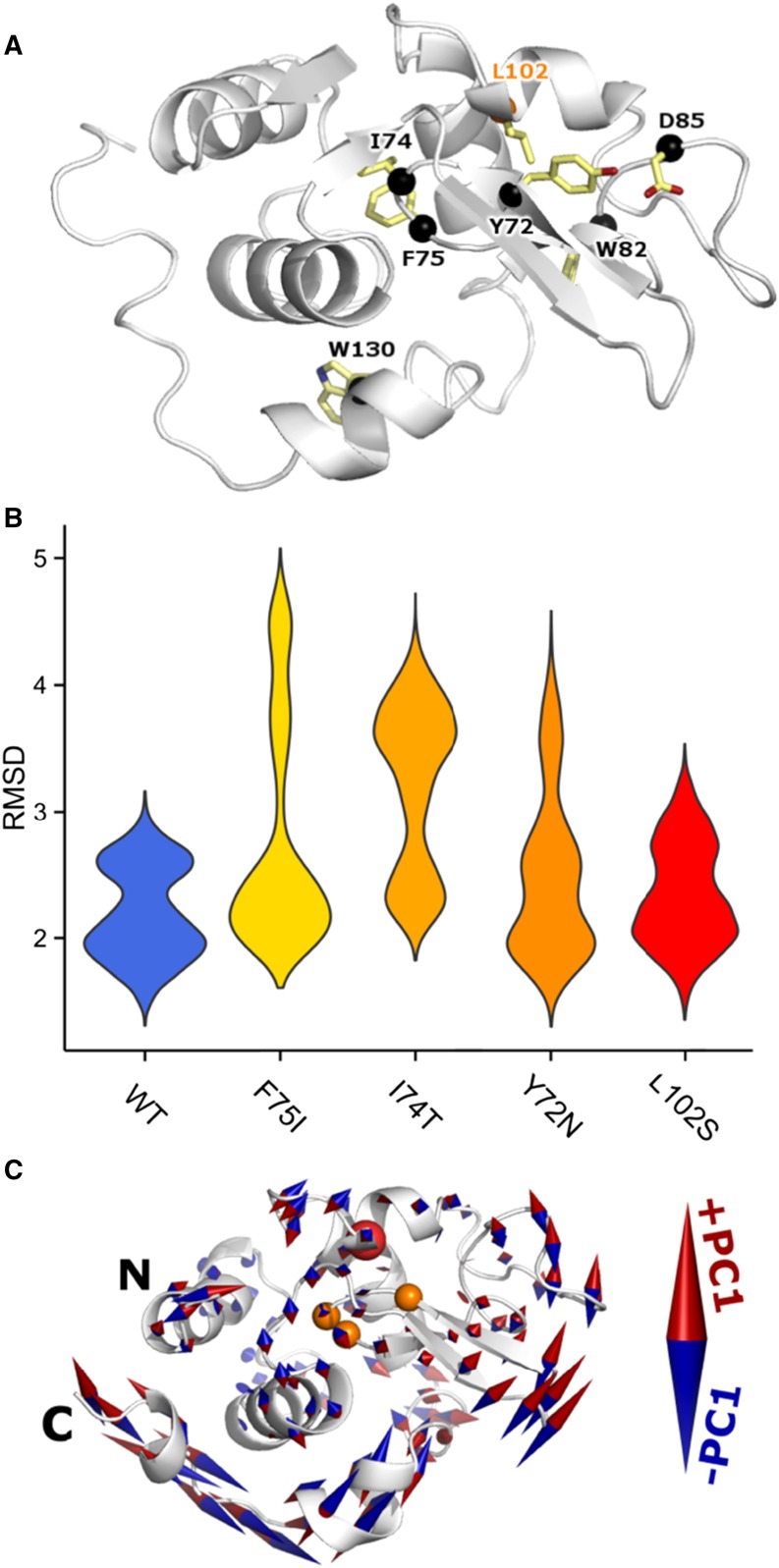
Lysozyme amyloidosis (ALys) is a rare form of hereditary amyloidosis that typically manifests with renal impairment, gastrointestinal (GI) symptoms, and sicca syndrome, whereas cardiac involvement is exceedingly rare and neuropathy has not been reported. Here, we describe a 40-year-old man with renal impairment, cardiac and GI symptoms, and peripheral neuropathy. Renal biopsy specimen analysis revealed amyloidosis with extensive involvement of glomeruli, vessels, and medulla. Amyloid was also detected in the GI tract. Echocardiographic and electrocardiographic findings were consistent with cardiac involvement. Proteomic analysis of Congo red-positive renal and GI amyloid deposits detected abundant lysozyme C protein. DNA sequencing of the lysozyme gene in the patient and his mother detected a heterozygous c.305T>C alteration in exon 3, which causes a leucine to serine substitution at codon 102 (Human Genome Variation Society nomenclature: p.Leu102Ser; legacy designation: L84S). We also detected the mutant peptide in the proband's renal and GI amyloid deposits. PolyPhen analysis predicted that the mutation damages the encoded protein. Molecular dynamics simulations suggested that the pathogenesis of ALys p.Leu102Ser is mediated by shifting the position of the central β-hairpin coordinated with an antiparallel motion of the C-terminal helix, which may alter the native-state structural ensemble of the molecule, leading to aggregation-prone intermediates.
Keywords: amyloidosis; hereditary amyloid; lysozyme; malfolding proteins.
Copyright © 2017 by the American Society of Nephrology.
Figures
References
-
- Dember LM: Amyloidosis-associated kidney disease. J Am Soc Nephrol 17: 3458–3471, 2006 - PubMed
-
- Valleix S, Verona G, Jourde-Chiche N, Nédelec B, Mangione PP, Bridoux F, Mangé A, Dogan A, Goujon JM, Lhomme M, Dauteuille C, Chabert M, Porcari R, Waudby CA, Relini A, Talmud PJ, Kovrov O, Olivecrona G, Stoppini M, Christodoulou J, Hawkins PN, Grateau G, Delpech M, Kontush A, Gillmore JD, Kalopissis AD, Bellotti V: D25V apolipoprotein C-III variant causes dominant hereditary systemic amyloidosis and confers cardiovascular protective lipoprotein profile. Nat Commun 7: 10353, 2016 - PMC - PubMed
-
- Nasr SH, Dasari S, Hasadsri L, Theis JD, Vrana JA, Gertz MA, Muppa P, Zimmermann MT, Grogg KL, Dispenzieri A, Sethi S, Highsmith WE Jr., Merlini G, Leung N, Kurtin PJ: Novel type of renal amyloidosis derived from Apolipoprotein-CII [published online ahead of print June 13, 2016]. J Am Soc Nephrol - PMC - PubMed
-
- Pepys MB, Hawkins PN, Booth DR, Vigushin DM, Tennent GA, Soutar AK, Totty N, Nguyen O, Blake CC, Terry CJ, Feest TG, Zalin AM, Hsuan JJ: Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature 362: 553–557, 1993 - PubMed
-
- Yazaki M, Farrell SA, Benson MD: A novel lysozyme mutation Phe57Ile associated with hereditary renal amyloidosis. Kidney Int 63: 1652–1657, 2003 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
