

Widespread Historical Contingency in Influenza Viruses
- PMID: 28049709
- PMCID: PMC5223518
- DOI: 10.1534/genetics.116.193979
Widespread Historical Contingency in Influenza Viruses
Abstract
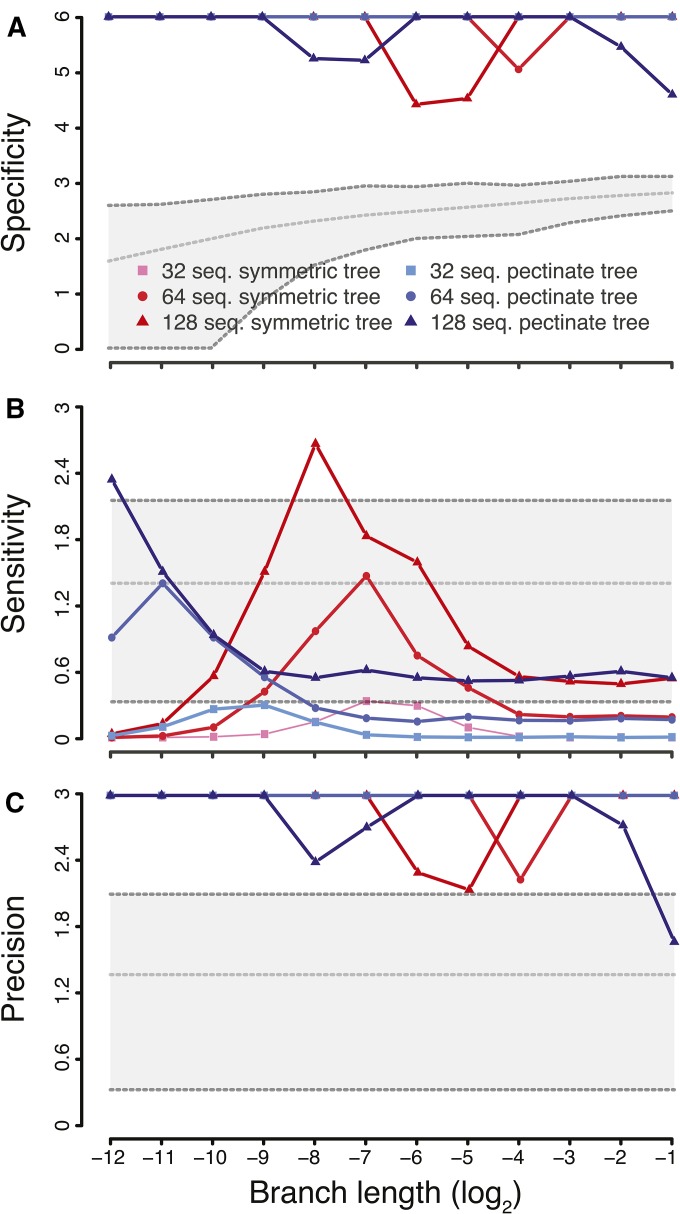
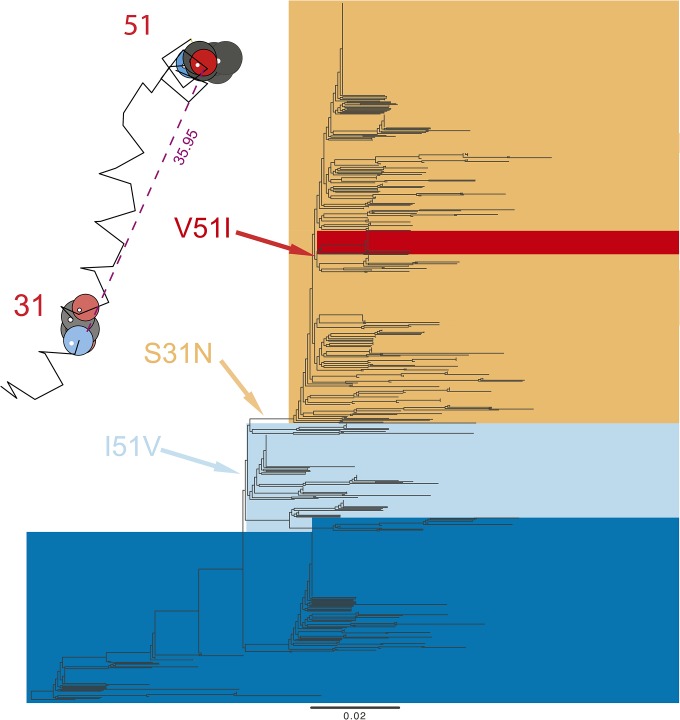
In systems biology and genomics, epistasis characterizes the impact that a substitution at a particular location in a genome can have on a substitution at another location. This phenomenon is often implicated in the evolution of drug resistance or to explain why particular "disease-causing" mutations do not have the same outcome in all individuals. Hence, uncovering these mutations and their locations in a genome is a central question in biology. However, epistasis is notoriously difficult to uncover, especially in fast-evolving organisms. Here, we present a novel statistical approach that replies on a model developed in ecology and that we adapt to analyze genetic data in fast-evolving systems such as the influenza A virus. We validate the approach using a two-pronged strategy: extensive simulations demonstrate a low-to-moderate sensitivity with excellent specificity and precision, while analyses of experimentally validated data recover known interactions, including in a eukaryotic system. We further evaluate the ability of our approach to detect correlated evolution during antigenic shifts or at the emergence of drug resistance. We show that in all cases, correlated evolution is prevalent in influenza A viruses, involving many pairs of sites linked together in chains; a hallmark of historical contingency. Strikingly, interacting sites are separated by large physical distances, which entails either long-range conformational changes or functional tradeoffs, for which we find support with the emergence of drug resistance. Our work paves a new way for the unbiased detection of epistasis in a wide range of organisms by performing whole-genome scans.
Keywords: correlated evolution; epistasis; influenza; networks.
Copyright © 2017 by the Genetics Society of America.
Figures



References
-
- Anisimova M., Gascuel O., 2006. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst. Biol. 55: 539–552. - PubMed
-
- Aris-Brosou S., Rodrigue N., 2012. The essentials of computational molecular evolution. Methods Mol. Biol. 855: 111–152. - PubMed
-
- Atchley W. R., Wollenberg K. R., Fitch W. M., Terhalle W., Dress A. W., 2000. Correlations among amino acid sites in bhlh protein domains: an information theoretic analysis. Mol. Biol. Evol. 17: 164–178. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
