

PaPrBaG: A machine learning approach for the detection of novel pathogens from NGS data
- PMID: 28051068
- PMCID: PMC5209729
- DOI: 10.1038/srep39194
PaPrBaG: A machine learning approach for the detection of novel pathogens from NGS data
Abstract
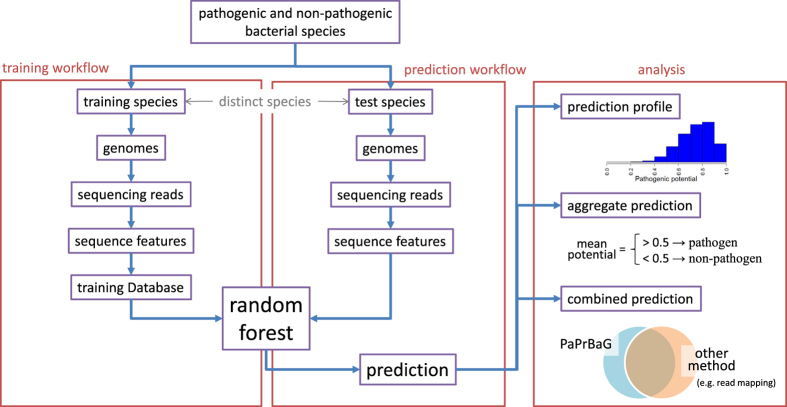
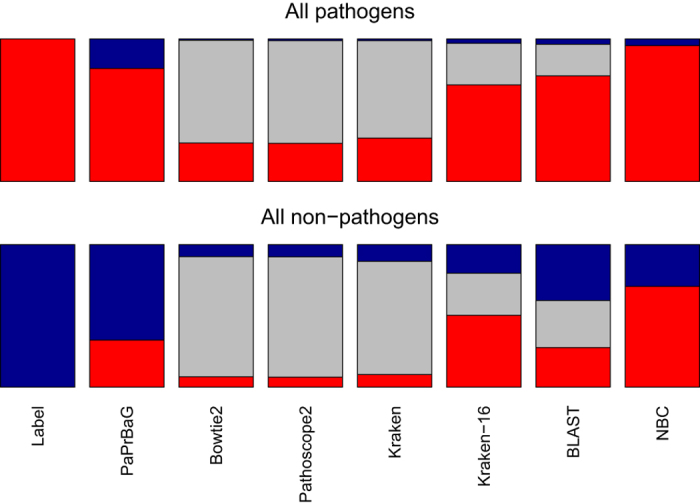
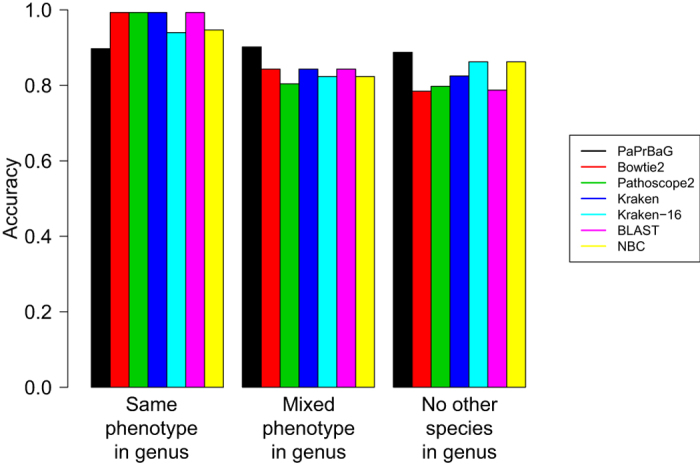
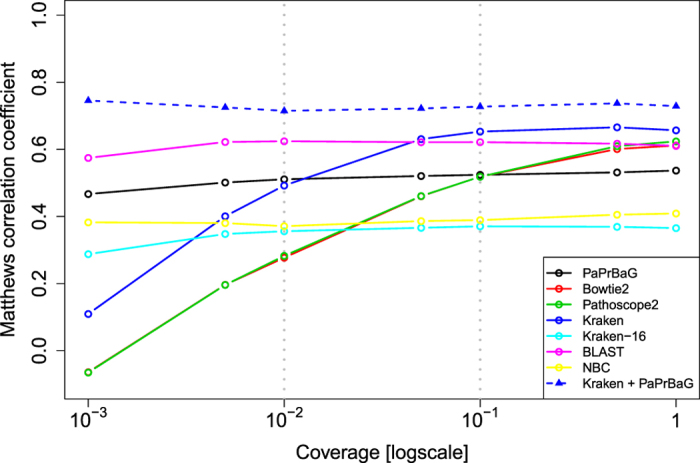
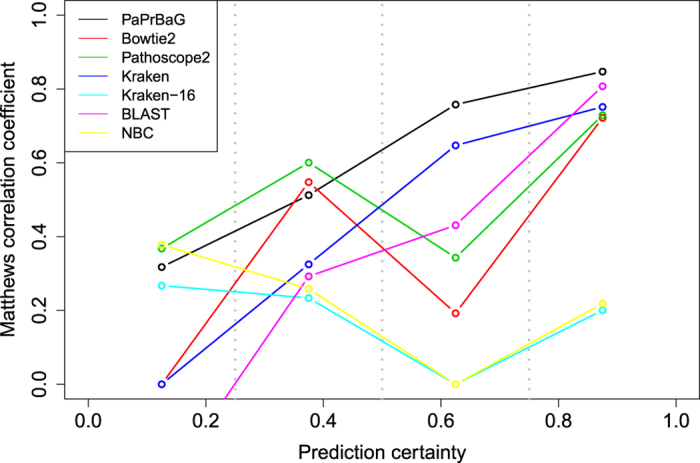
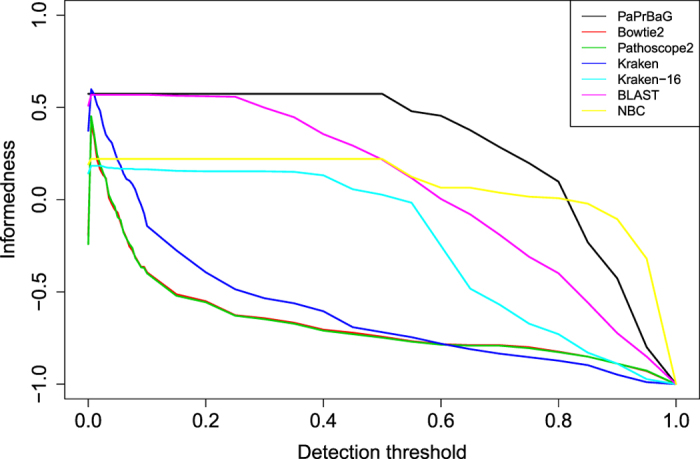
The reliable detection of novel bacterial pathogens from next-generation sequencing data is a key challenge for microbial diagnostics. Current computational tools usually rely on sequence similarity and often fail to detect novel species when closely related genomes are unavailable or missing from the reference database. Here we present the machine learning based approach PaPrBaG (Pathogenicity Prediction for Bacterial Genomes). PaPrBaG overcomes genetic divergence by training on a wide range of species with known pathogenicity phenotype. To that end we compiled a comprehensive list of pathogenic and non-pathogenic bacteria with human host, using various genome metadata in conjunction with a rule-based protocol. A detailed comparative study reveals that PaPrBaG has several advantages over sequence similarity approaches. Most importantly, it always provides a prediction whereas other approaches discard a large number of sequencing reads with low similarity to currently known reference genomes. Furthermore, PaPrBaG remains reliable even at very low genomic coverages. CombiningPaPrBaG with existing approaches further improves prediction results.
Figures
References
-
- Juhas M. Horizontal gene transfer in human pathogens. Critical Reviews in Microbiology 41, 101–108 (2015). - PubMed
-
- Merhej V., Georgiades K. & Raoult D. Postgenomic analysis of bacterial pathogens repertoire reveals genome reduction rather than virulence factors. Briefings in Functional Genomics 12, 291–304 (2013). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
