

Domain Organization and Evolution of the Highly Divergent 5' Coding Region of Genomes of Arteriviruses, Including the Novel Possum Nidovirus
- PMID: 28053107
- PMCID: PMC5331827
- DOI: 10.1128/JVI.02096-16
Domain Organization and Evolution of the Highly Divergent 5' Coding Region of Genomes of Arteriviruses, Including the Novel Possum Nidovirus
Abstract
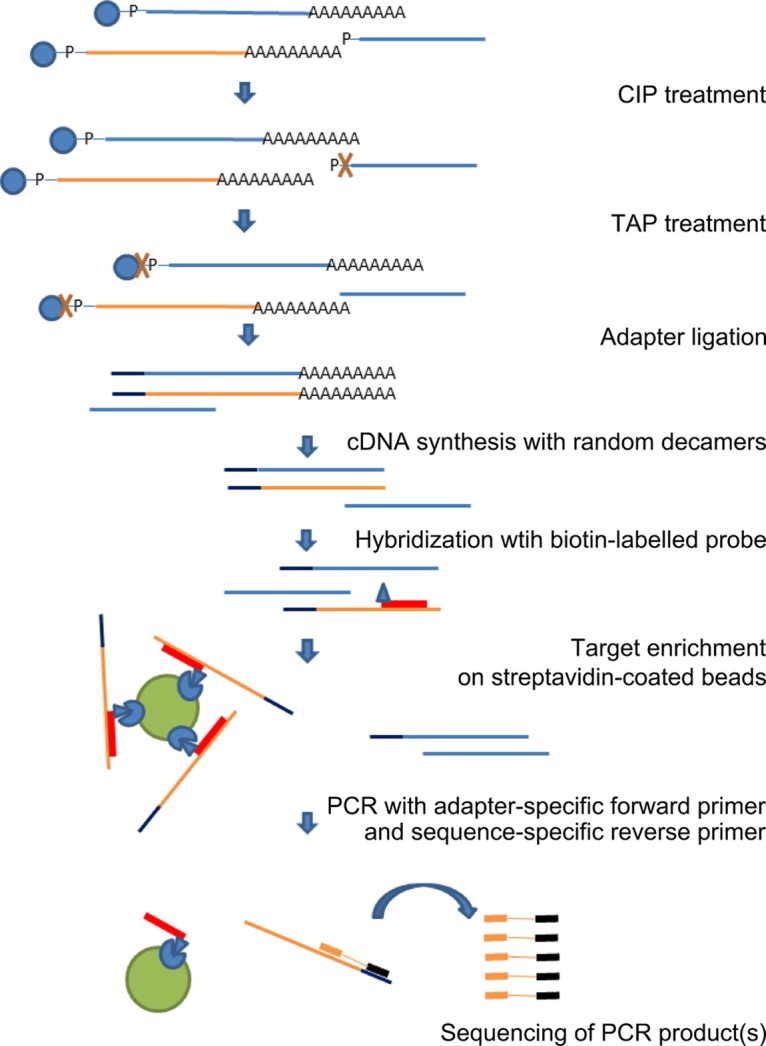
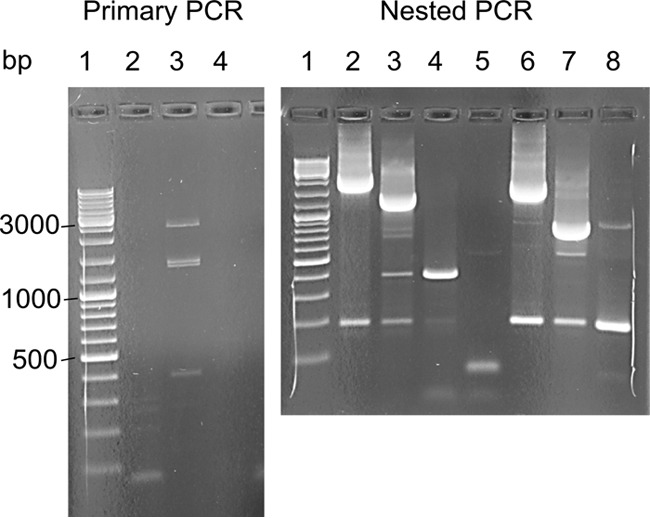
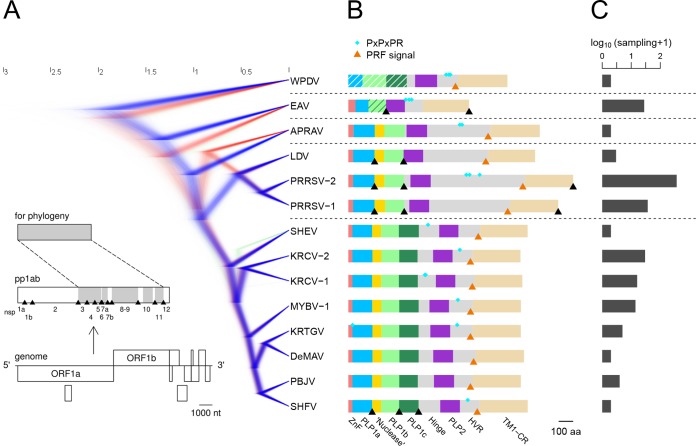
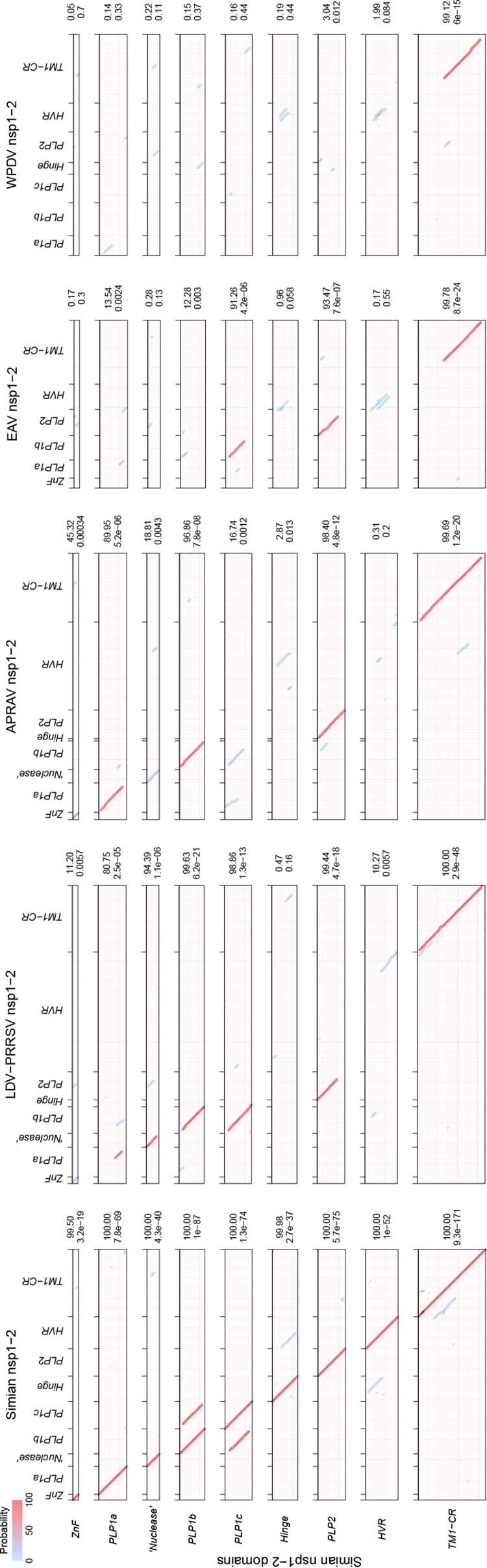
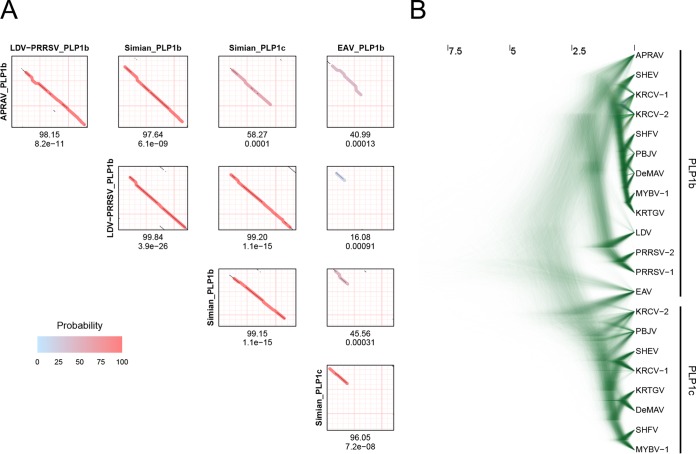
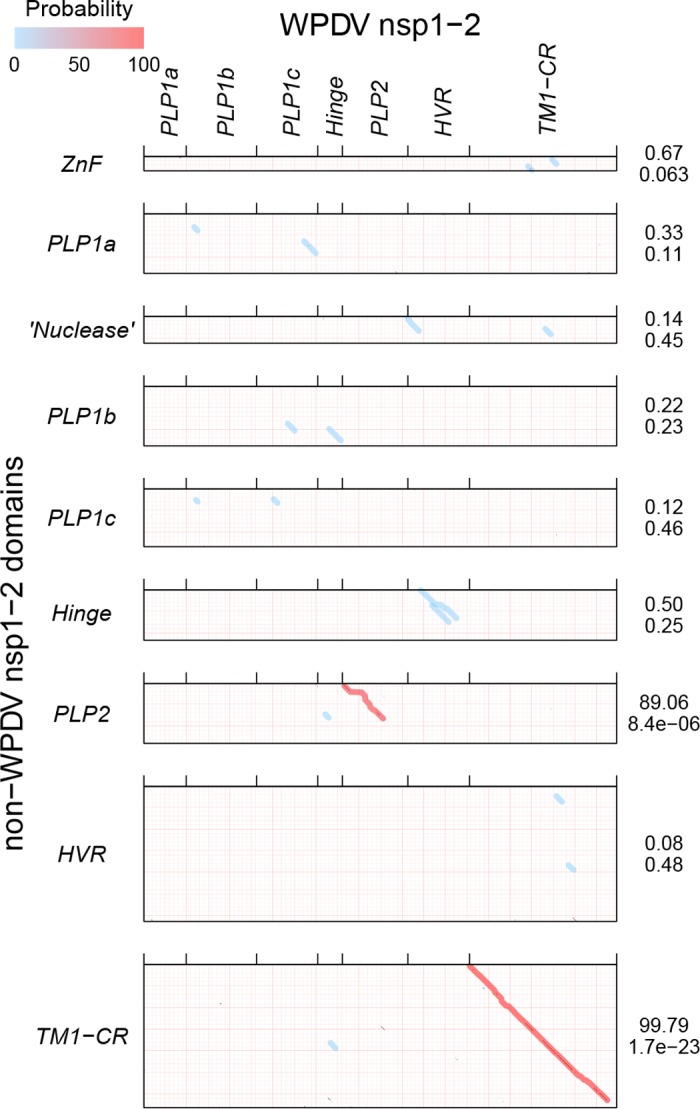
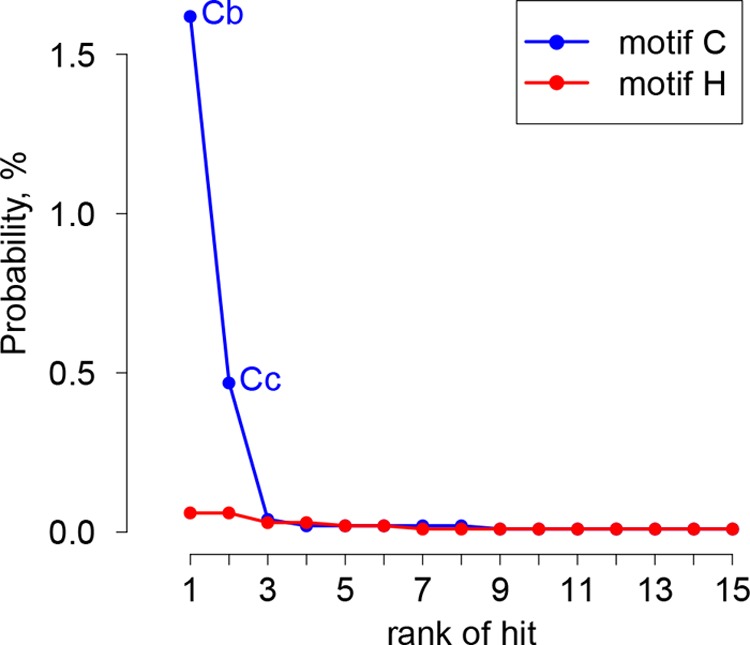
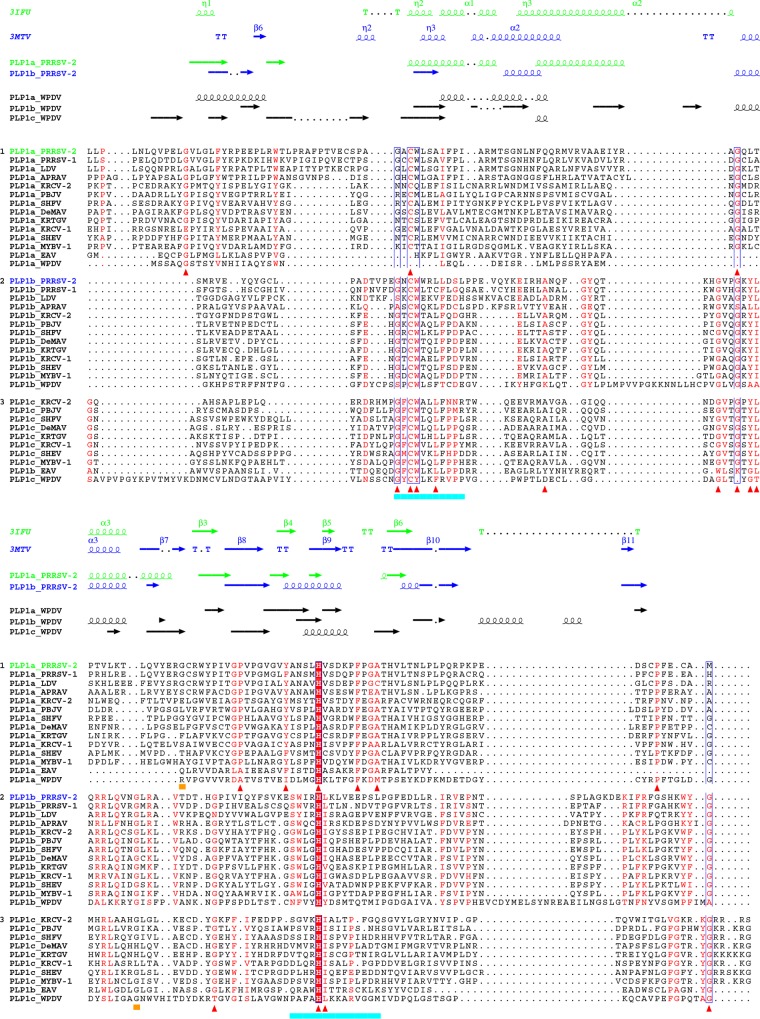
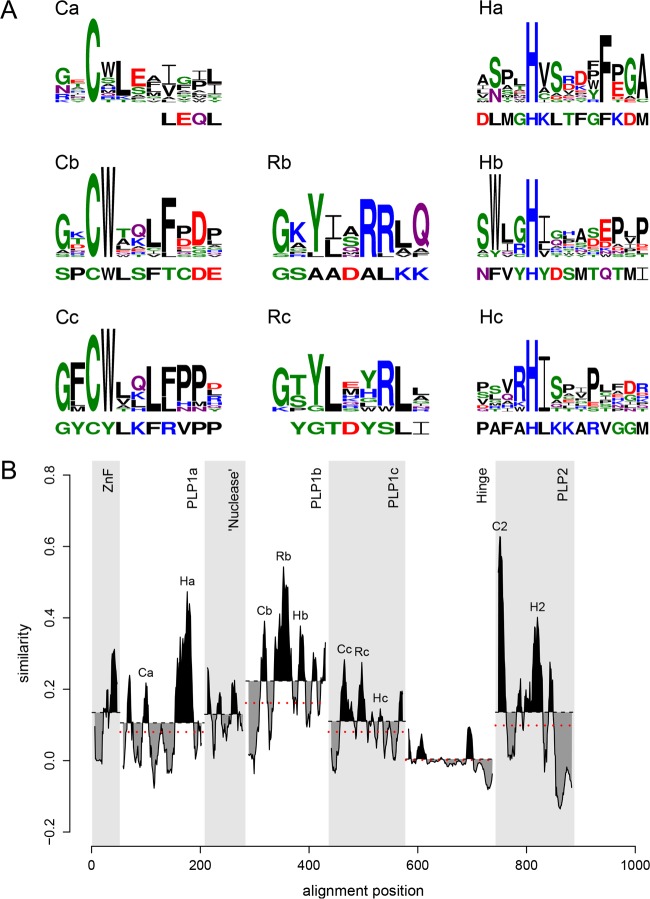
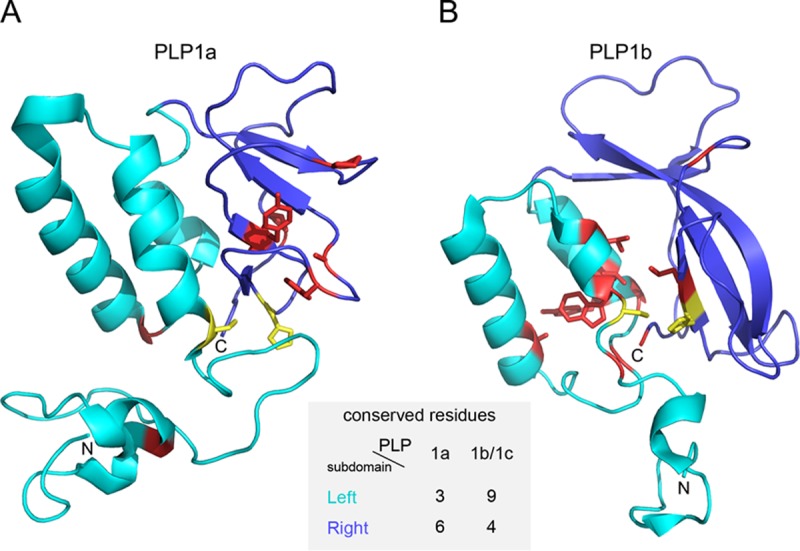
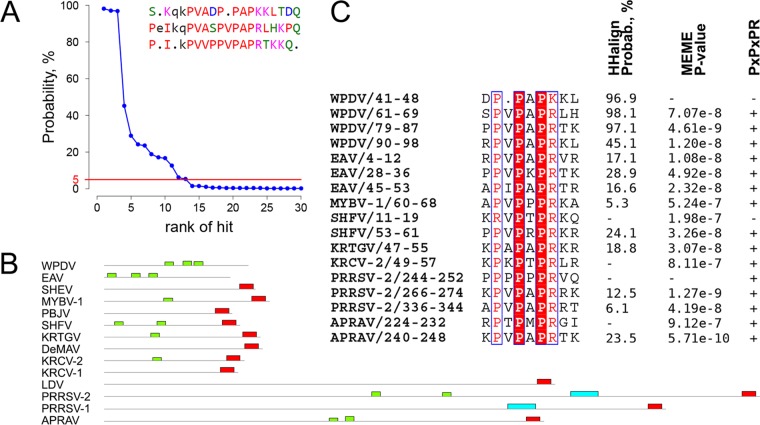
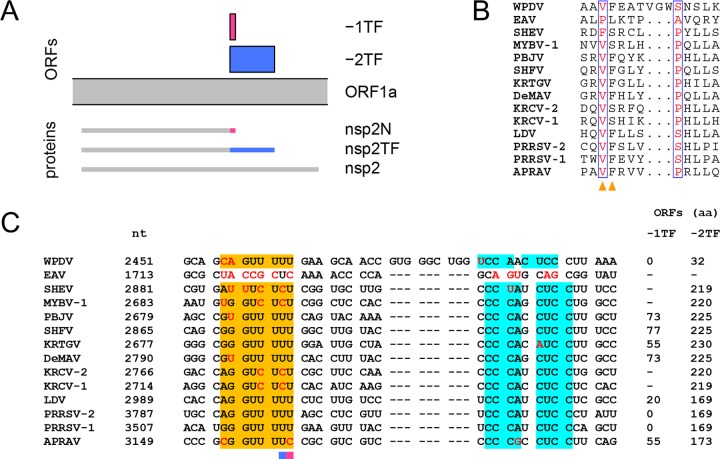
In five experimentally characterized arterivirus species, the 5'-end genome coding region encodes the most divergent nonstructural proteins (nsp's), nsp1 and nsp2, which include papain-like proteases (PLPs) and other poorly characterized domains. These are involved in regulation of transcription, polyprotein processing, and virus-host interaction. Here we present results of a bioinformatics analysis of this region of 14 arterivirus species, including that of the most distantly related virus, wobbly possum disease virus (WPDV), determined by a modified 5' rapid amplification of cDNA ends (RACE) protocol. By combining profile-profile comparisons and phylogeny reconstruction, we identified an association of the four distinct domain layouts of nsp1-nsp2 with major phylogenetic lineages, implicating domain gain, including duplication, and loss in the early nsp1 evolution. Specifically, WPDV encodes highly divergent homologs of PLP1a, PLP1b, PLP1c, and PLP2, with PLP1a lacking the catalytic Cys residue, but does not encode nsp1 Zn finger (ZnF) and "nuclease" domains, which are conserved in other arteriviruses. Unexpectedly, our analysis revealed that the only catalytically active nsp1 PLP of equine arteritis virus (EAV), known as PLP1b, is most similar to PLP1c and thus is likely to be a PLP1b paralog. In all non-WPDV arteriviruses, PLP1b/c and PLP1a show contrasting patterns of conservation, with the N- and C-terminal subdomains, respectively, being enriched with conserved residues, which is indicative of different functional specializations. The least conserved domain of nsp2, the hypervariable region (HVR), has its size varied 5-fold and includes up to four copies of a novel PxPxPR motif that is potentially recognized by SH3 domain-containing proteins. Apparently, only EAV lacks the signal that directs -2 ribosomal frameshifting in the nsp2 coding region.IMPORTANCE Arteriviruses comprise a family of mammalian enveloped positive-strand RNA viruses that include some of the most economically important pathogens of swine. Most of our knowledge about this family has been obtained through characterization of viruses from five species: Equine arteritis virus, Simian hemorrhagic fever virus, Lactate dehydrogenase-elevating virus, Porcine respiratory and reproductive syndrome virus 1, and Porcine respiratory and reproductive syndrome virus 2 Here we present the results of comparative genomics analyses of viruses from all known 14 arterivirus species, including the most distantly related virus, WPDV, whose genome sequence was completed in this study. Our analysis focused on the multifunctional 5'-end genome coding region that encodes multidomain nonstructural proteins 1 and 2. Using diverse bioinformatics techniques, we identified many patterns of evolutionary conservation that are specific to members of distinct arterivirus species, both characterized and novel, or their groups. They are likely associated with structural and functional determinants important for virus replication and virus-host interaction.
Keywords: arterivirus; bioinformatics; comparative genomics; duplication; evolution; nidovirus; papain-like proteases; phylogenetic analysis; ribosome frameshifting; wobbly possum disease.
Copyright © 2017 American Society for Microbiology.
Figures



References
-
- Faaberg KS, Balasuriya UB, Brinton MA, Gorbalenya AE, Leung FC-C, Nauwynck H, Snijder EJ, Stadejek T, Yang H, Yoo D. 2012. Family Arteriviridae, p 796–805. In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed), Virus taxonomy: classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses. Academic Press, London, United Kingdom.
-
- Adams MJ, Lefkowitz EJ, King AM, Harrach B, Harrison RL, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Mushegian AR, Nibert M, Sabanadzovic S, Sanfacon H, Siddell SG, Simmonds P, Varsani A, Zerbini FM, Gorbalenya AE, Davison AJ. 2016. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Arch Virol 161:2921–2949. doi: 10.1007/s00705-016-2977-6. - DOI - PMC - PubMed
-
- Balasuriya UB, Snijder EJ, Heidner HW, Zhang J, Zevenhoven-Dobbe JC, Boone JD, McCollum WH, Timoney PJ, MacLachlan NJ. 2007. Development and characterization of an infectious cDNA clone of the virulent Bucyrus strain of equine arteritis virus. J Gen Virol 88:918–924. doi: 10.1099/vir.0.82415-0. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
