

Gene profile of fibroblasts identify relation of CCL8 with idiopathic pulmonary fibrosis
- PMID: 28057004
- PMCID: PMC5216573
- DOI: 10.1186/s12931-016-0493-6
Gene profile of fibroblasts identify relation of CCL8 with idiopathic pulmonary fibrosis
Abstract
Background: Idiopathic pulmonary fibrosis (IPF) is characterized by the complex interaction of cells involved in chronic inflammation and fibrosis. Global gene expression of a homogenous cell population will identify novel candidate genes.
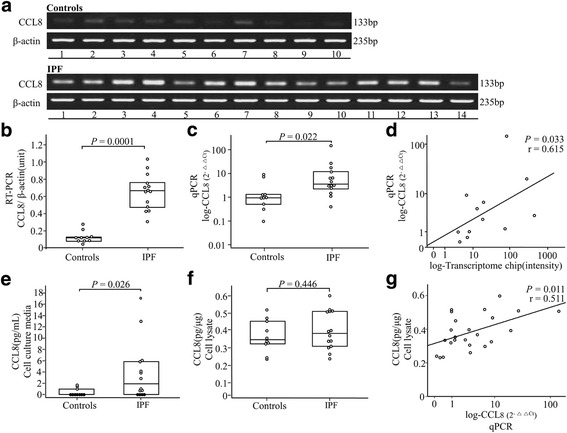
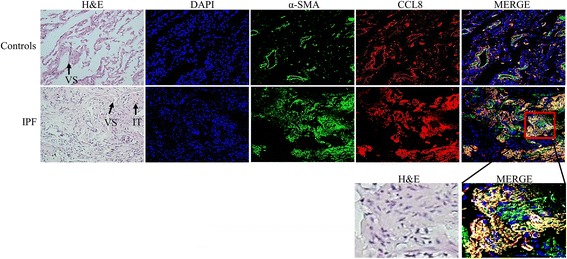
Methods: Gene expression of fibroblasts derived from lung tissues (8 IPF and 4 controls) was profiled, and ontology and functional pathway were analyzed in the genes exhibiting >2 absolute fold changes with p-values < 0.05. CCL8 mRNA and protein levels were quantified using real-time PCR and ELISA. CCL8 localization was evaluated by immunofluorescence staining.
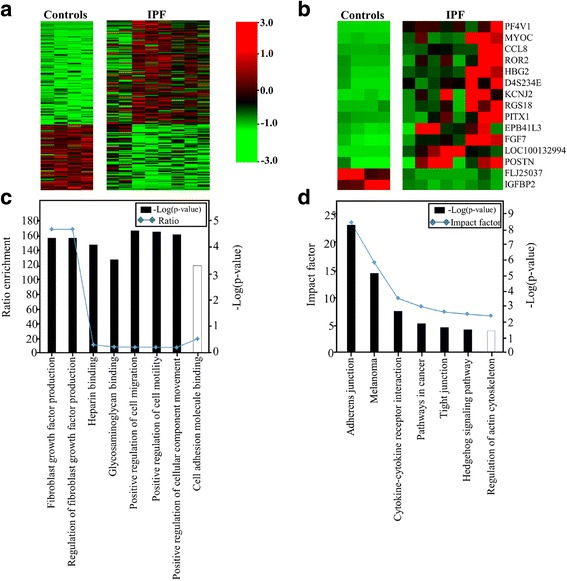
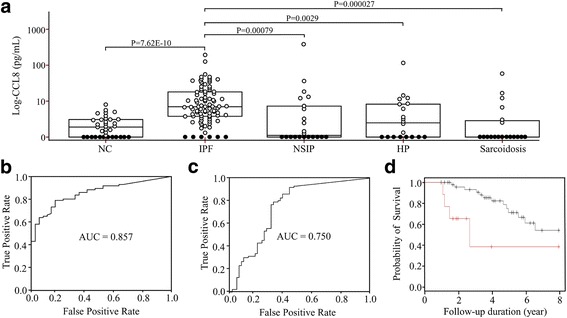
Results: One hundred seventy eight genes differentially expressed and 15 genes exhibited >10-fold change. Among them, 13 were novel in relation with IPF. CCL8 expression was 22.8-fold higher in IPF fibroblasts. The levels of CCL8 mRNA and protein were 3 and 9-fold higher in 14 IPF fibroblasts than those in 10 control fibroblasts by real-time PCR and ELISA (p = 0.022 and p = 0.026, respectively). The CCL8 concentrations in BAL fluid was significantly higher in 86 patients with IPF than those in 41 controls, and other interstitial lung diseases including non-specific interstitial pneumonia (n = 22), hypersensitivity pneumonitis (n = 20) and sarcoidosis (n = 19) (p < 0.005, respectively). Cut-off values of 2.29 pg/mL and 0.43 pg/mL possessed 80.2 and 70.7% accuracy for the discrimination of IPF from NC and the other lung diseases, respectively. IPF subjects with CCL8 levels >28.61 pg/mL showed shorter survival compared to those with lower levels (p = 0.012). CCL8 was expressed by α-SMA-positive cells in the interstitium of IPF.
Conclusions: Transcriptome analysis identified several novel IPF-related genes. Among them, CCL8 is a candidate molecule for the differential diagnosis and prediction of survival.
Keywords: CCL8; Gene expression; IPF; Transcriptome.
Figures
References
-
- Zuo F, Kaminski N, Eugui E, Allard J, Yakhini Z, Ben-Dor A, Lollini L, Morris D, Kim Y, DeLustro B, et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci U S A. 2002;99:6292–7. doi: 10.1073/pnas.092134099. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
