

GTB - an online genome tolerance browser
- PMID: 28061747
- PMCID: PMC5219737
- DOI: 10.1186/s12859-016-1436-4
GTB - an online genome tolerance browser
Abstract
Background: Accurate methods capable of predicting the impact of single nucleotide variants (SNVs) are assuming ever increasing importance. There exists a plethora of in silico algorithms designed to help identify and prioritize SNVs across the human genome for further investigation. However, no tool exists to visualize the predicted tolerance of the genome to mutation, or the similarities between these methods.
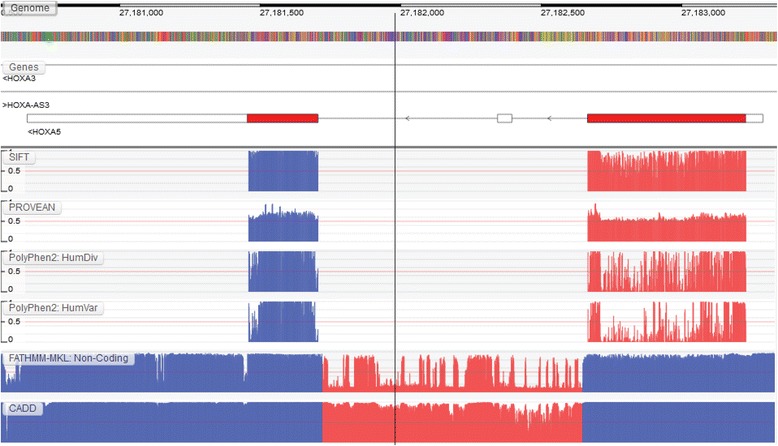
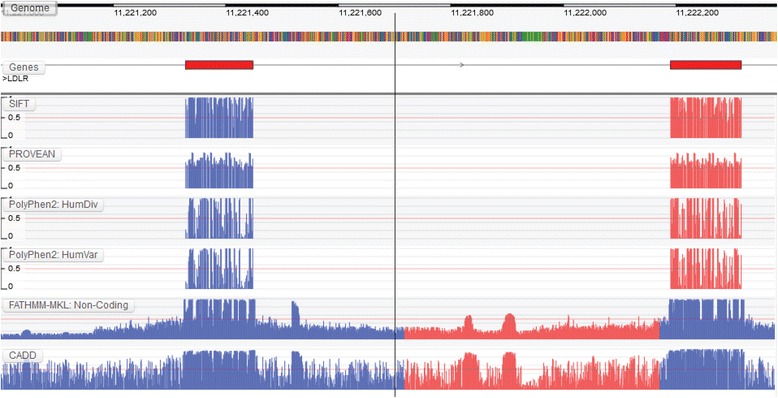
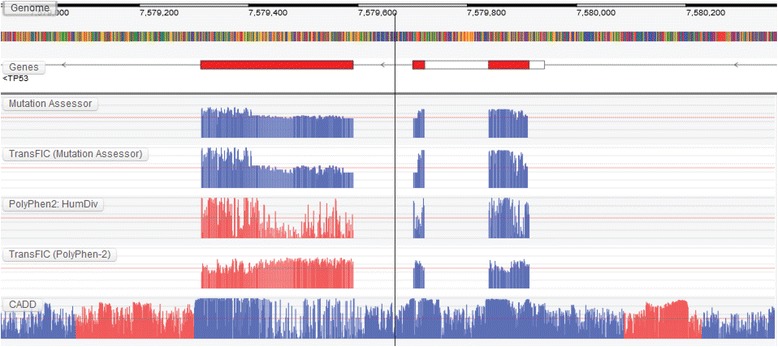
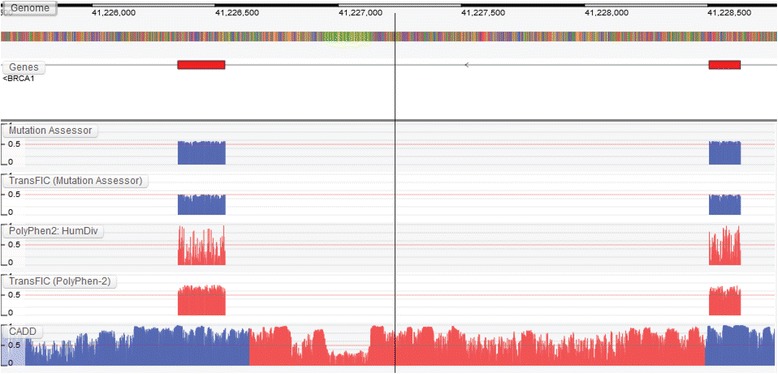
Results: We present the Genome Tolerance Browser (GTB, http://gtb.biocompute.org.uk ): an online genome browser for visualizing the predicted tolerance of the genome to mutation. The server summarizes several in silico prediction algorithms and conservation scores: including 13 genome-wide prediction algorithms and conservation scores, 12 non-synonymous prediction algorithms and four cancer-specific algorithms.
Conclusion: The GTB enables users to visualize the similarities and differences between several prediction algorithms and to upload their own data as additional tracks; thereby facilitating the rapid identification of potential regions of interest.
Keywords: Genome browser; Genome tolerance; Mutation; Pathogenicity prediction; Prediction algorithm; SNVs; Variant effect prediction.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
