

treeman: an R package for efficient and intuitive manipulation of phylogenetic trees
- PMID: 28061884
- PMCID: PMC5219798
- DOI: 10.1186/s13104-016-2340-8
treeman: an R package for efficient and intuitive manipulation of phylogenetic trees
Abstract
Background: Phylogenetic trees are hierarchical structures used for representing the inter-relationships between biological entities. They are the most common tool for representing evolution and are essential to a range of fields across the life sciences. The manipulation of phylogenetic trees-in terms of adding or removing tips-is often performed by researchers not just for reasons of management but also for performing simulations in order to understand the processes of evolution. Despite this, the most common programming language among biologists, R, has few class structures well suited to these tasks.
Results: We present an R package that contains a new class, called TreeMan, for representing the phylogenetic tree. This class has a list structure allowing phylogenetic trees to be manipulated more efficiently. Computational running times are reduced because of the ready ability to vectorise and parallelise methods. Development is also improved due to fewer lines of code being required for performing manipulation processes.
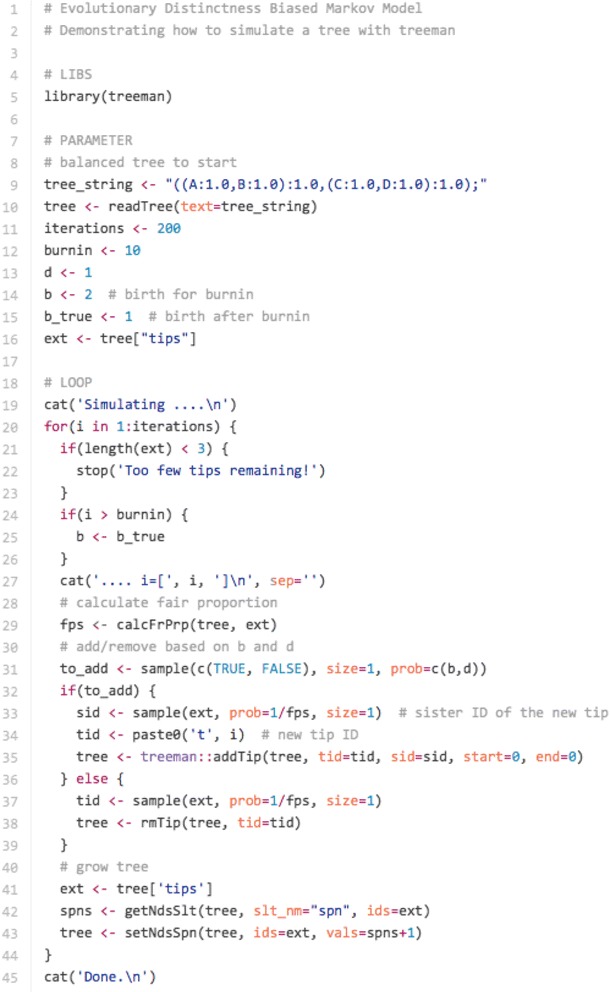
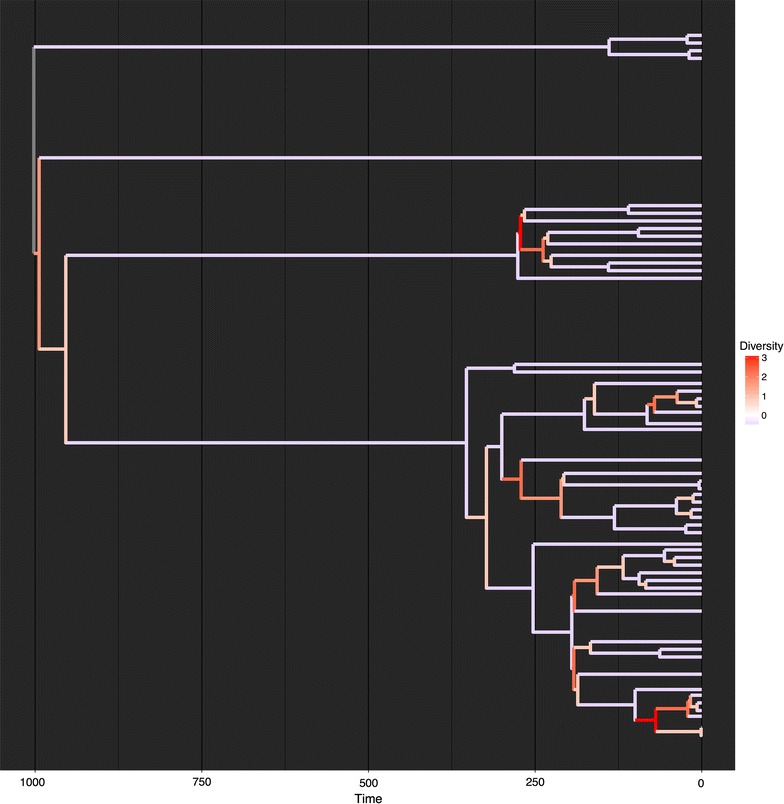
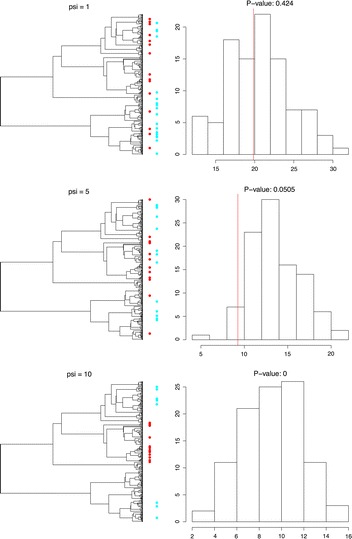
Conclusions: We present three use cases-pinning missing taxa to a supertree, simulating evolution with a tree-growth model and detecting significant phylogenetic turnover-that demonstrate the new package's speed and simplicity.
Keywords: Evolution; Phylogenetic trees; R; Statistical computing; Tree simulation.
Figures
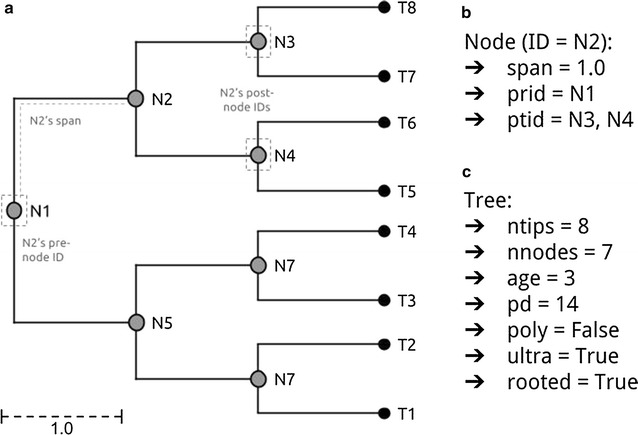
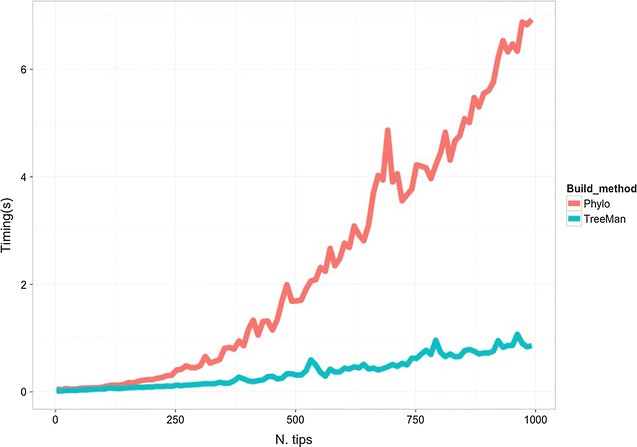
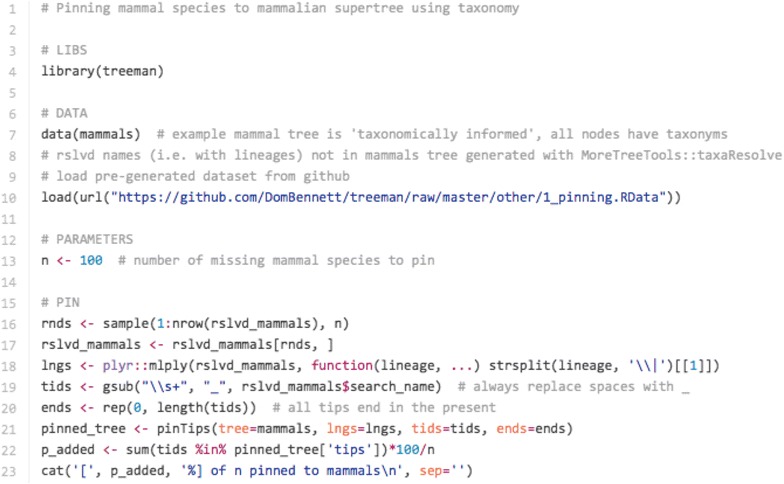
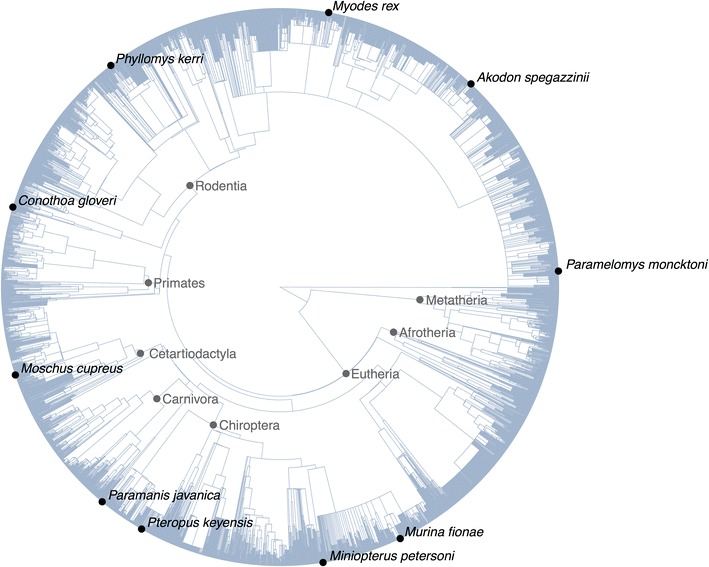

References
-
- Paradis E, Blomberg S, Bolker B, Claude J, Cuong HS, Desper R, Didier G, Durand B, Dutheil J, Gascuel O. ape: Analyses of phylogenetics and evolution. 2016. https://cran.r-project.org/web/packages/ape/index.html. Accessed 24 Feb 2016.
-
- Revell LJ. phytools: an R package for phylogenetic comparative biology (and other things) Methods Ecol Evol. 2012;3:217–223. doi: 10.1111/j.2041-210X.2011.00169.x. - DOI
-
- Michonneau F. phylobase: Base package for phylogenetic structures and comparative data. 2016. https://cran.r-project.org/web/packages/phylobase/index.html. Accessed 3 Sep 2016.
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
