

Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma
- PMID: 28064239
- PMCID: PMC5270390
- DOI: 10.1182/blood-2016-07-729954
Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma
Abstract
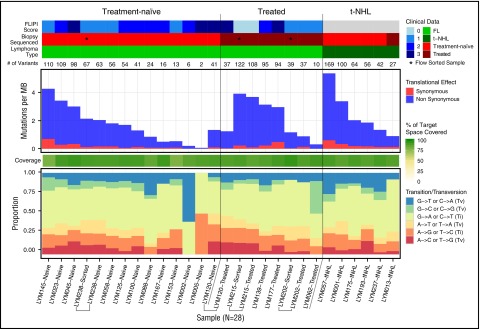
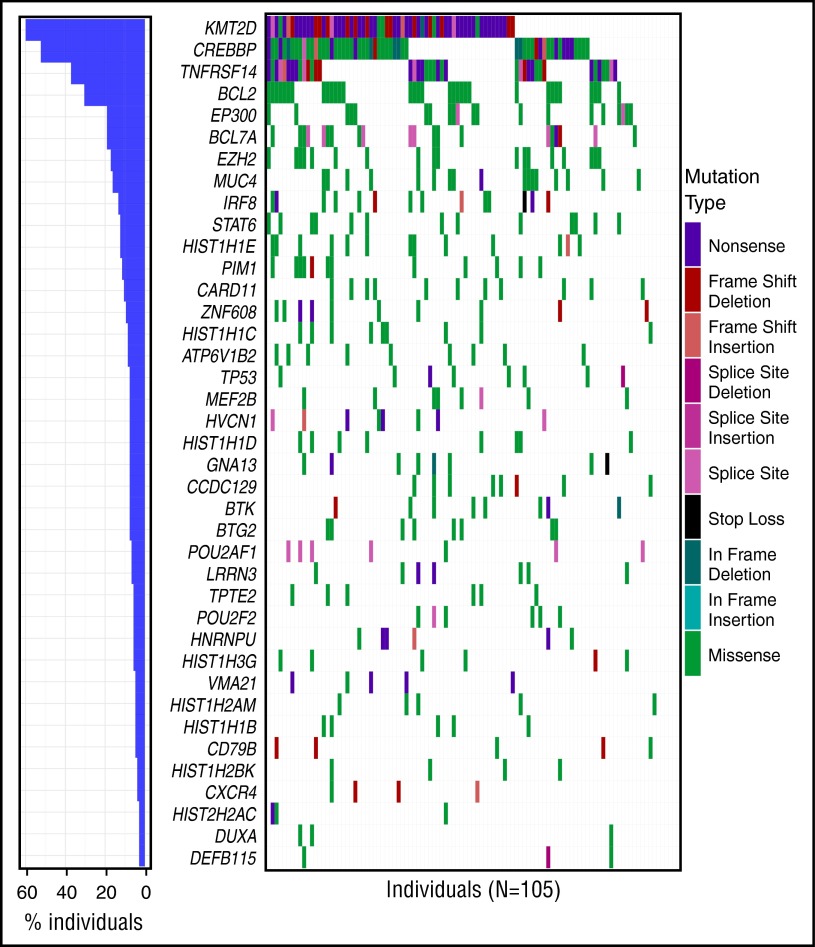
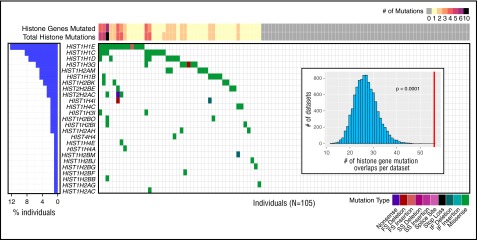
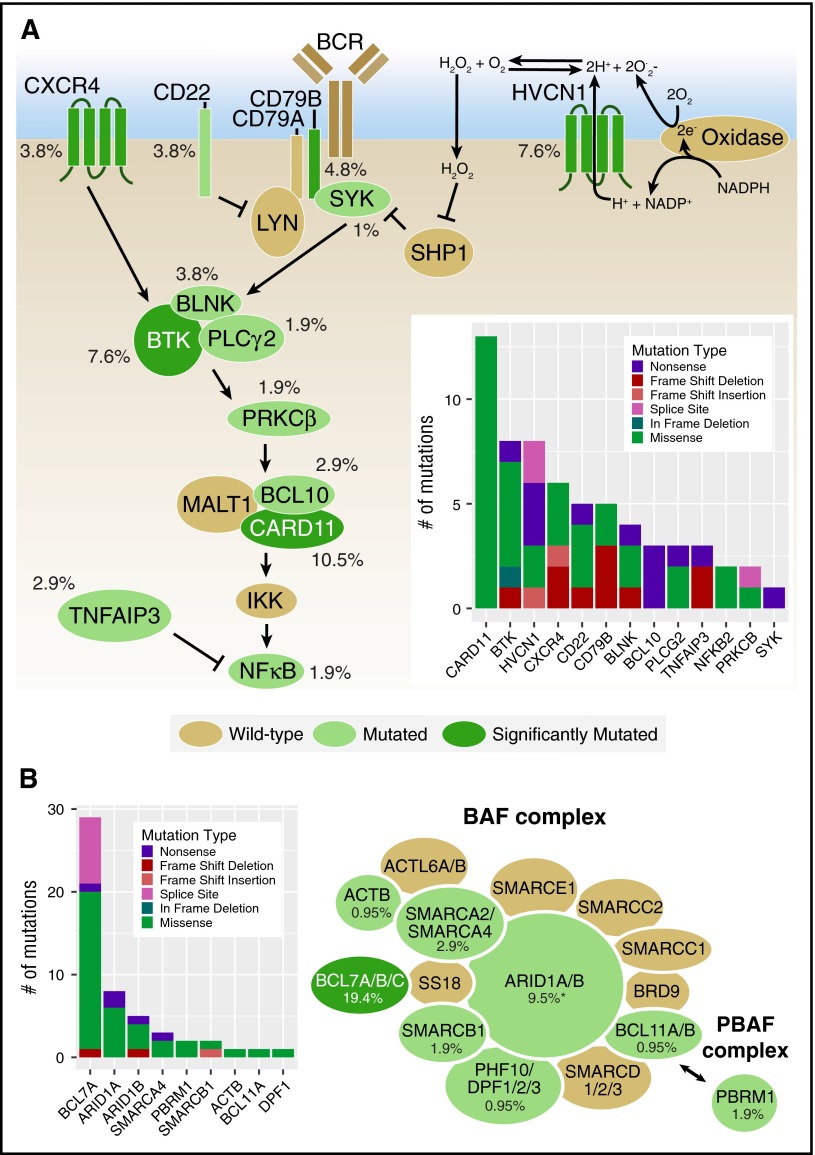
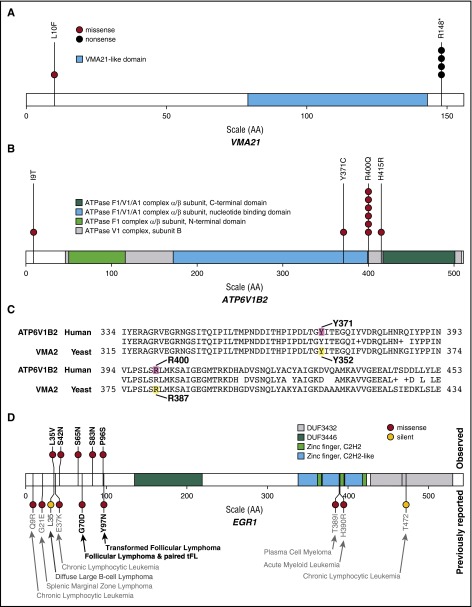
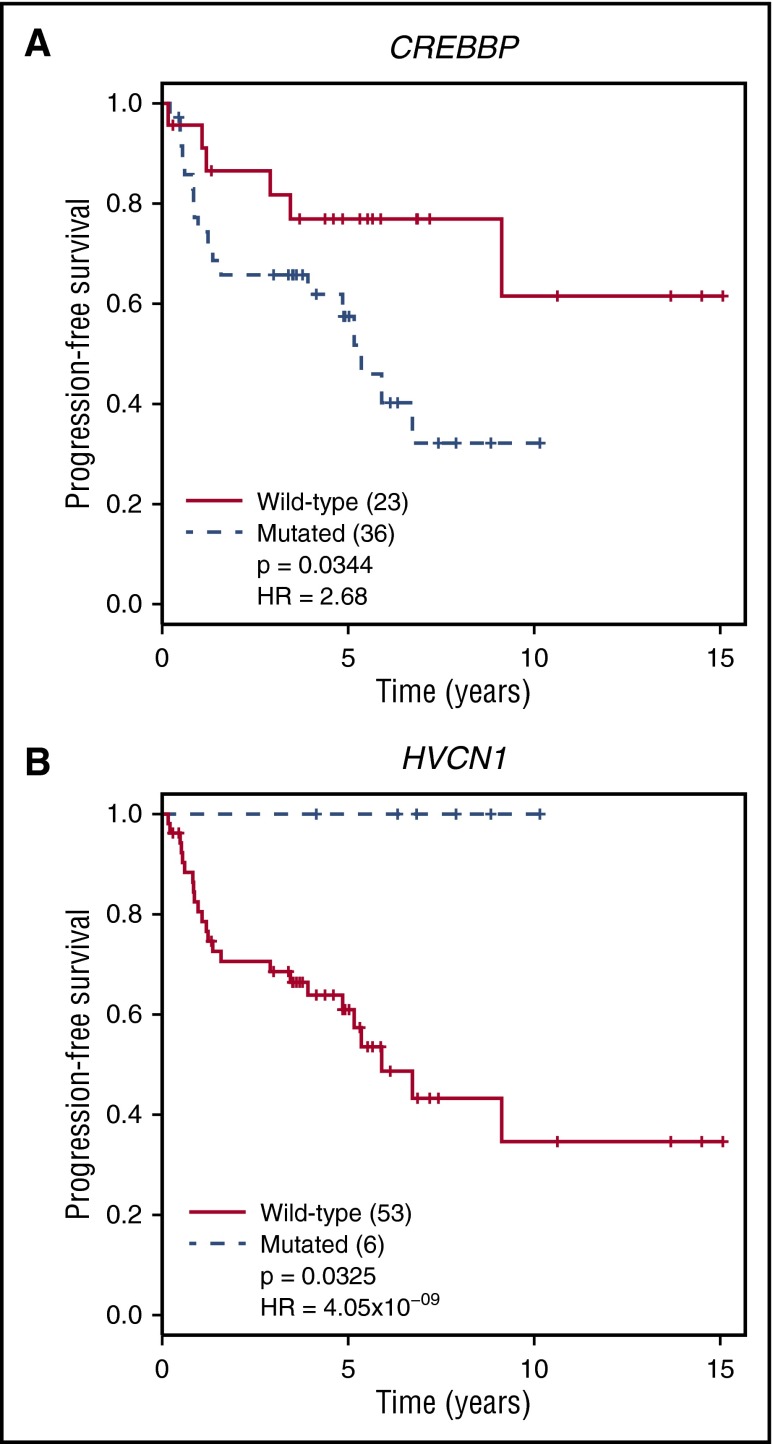
Follicular lymphoma (FL) is the most common form of indolent non-Hodgkin lymphoma, yet it remains only partially characterized at the genomic level. To improve our understanding of the genetic underpinnings of this incurable and clinically heterogeneous disease, whole-exome sequencing was performed on tumor/normal pairs from a discovery cohort of 24 patients with FL. Using these data and mutations identified in other B-cell malignancies, 1716 genes were sequenced in 113 FL tumor samples from 105 primarily treatment-naive individuals. We identified 39 genes that were mutated significantly above background mutation rates. CREBBP mutations were associated with inferior PFS. In contrast, mutations in previously unreported HVCN1, a voltage-gated proton channel-encoding gene and B-cell receptor signaling modulator, were associated with improved PFS. In total, 47 (44.8%) patients harbor mutations in the interconnected B-cell receptor (BCR) and CXCR4 signaling pathways. Histone gene mutations were more frequent than previously reported (identified in 43.8% of patients) and often co-occurred (17.1% of patients). A novel, recurrent hotspot was identified at a posttranslationally modified residue in the histone H2B family. This study expands the number of mutated genes described in several known signaling pathways and complexes involved in lymphoma pathogenesis (BCR, Notch, SWitch/sucrose nonfermentable (SWI/SNF), vacuolar ATPases) and identified novel recurrent mutations (EGR1/2, POU2AF1, BTK, ZNF608, HVCN1) that require further investigation in the context of FL biology, prognosis, and treatment.
© 2017 by The American Society of Hematology.
Figures
Comment in
-
Recurrent mutations and targeted therapy in FL.Blood. 2017 Jan 26;129(4):402-403. doi: 10.1182/blood-2016-12-753145. Blood. 2017. PMID: 28126956 No abstract available.
References
-
- Nabhan C, Aschebrook-Kilfoy B, Chiu BC, Kruczek K, Smith SM, Evens AM. The impact of race, age, and sex in follicular lymphoma: A comprehensive SEER analysis across consecutive treatment eras. Am J Hematol. 2014;89(6):633-638. - PubMed
-
- Jacobson CA, Freedman AS. Is observation dead in follicular lymphoma? Still appropriate. J Natl Compr Canc Netw. 2015;13(3):367-370. - PubMed
-
- Kahl BS, Yang DT. Follicular lymphoma: evolving therapeutic strategies. Blood. 2016;127(17):2055-2063. - PubMed
-
- Casulo C, Burack WR, Friedberg JW. Transformed follicular non-Hodgkin lymphoma. Blood. 2015;125(1):40-47. - PubMed
-
- Basso K, Dalla-Favera R. Germinal centres and B cell lymphomagenesis. Nat Rev Immunol. 2015;15(3):172-184. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
