

Mutations in ATP6V1E1 or ATP6V1A Cause Autosomal-Recessive Cutis Laxa
- PMID: 28065471
- PMCID: PMC5294668
- DOI: 10.1016/j.ajhg.2016.12.010
Mutations in ATP6V1E1 or ATP6V1A Cause Autosomal-Recessive Cutis Laxa
Erratum in
-
Mutations in ATP6V1E1 or ATP6V1A Cause Autosomal-Recessive Cutis Laxa.Am J Hum Genet. 2020 Aug 6;107(2):374. doi: 10.1016/j.ajhg.2020.07.013. Am J Hum Genet. 2020. PMID: 32763190 Free PMC article. No abstract available.
Abstract
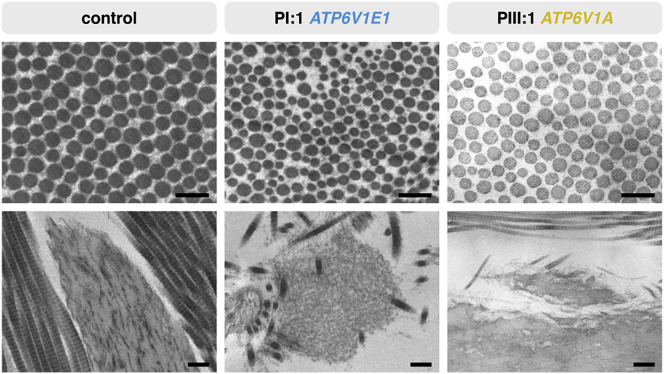
Defects of the V-type proton (H+) ATPase (V-ATPase) impair acidification and intracellular trafficking of membrane-enclosed compartments, including secretory granules, endosomes, and lysosomes. Whole-exome sequencing in five families affected by mild to severe cutis laxa, dysmorphic facial features, and cardiopulmonary involvement identified biallelic missense mutations in ATP6V1E1 and ATP6V1A, which encode the E1 and A subunits, respectively, of the V1 domain of the heteromultimeric V-ATPase complex. Structural modeling indicated that all substitutions affect critical residues and inter- or intrasubunit interactions. Furthermore, complexome profiling, a method combining blue-native gel electrophoresis and liquid chromatography tandem mass spectrometry, showed that they disturb either the assembly or the stability of the V-ATPase complex. Protein glycosylation was variably affected. Abnormal vesicular trafficking was evidenced by delayed retrograde transport after brefeldin A treatment and abnormal swelling and fragmentation of the Golgi apparatus. In addition to showing reduced and fragmented elastic fibers, the histopathological hallmark of cutis laxa, transmission electron microscopy of the dermis also showed pronounced changes in the structure and organization of the collagen fibers. Our findings expand the clinical and molecular spectrum of metabolic cutis laxa syndromes and further link defective extracellular matrix assembly to faulty protein processing and cellular trafficking caused by genetic defects in the V-ATPase complex.
Keywords: ARCL2; ATP6V1A; ATP6V1E1; CDG; Golgi apparatus; V-ATPase; autosomal recessive; cellular trafficking; congenital disorder of glycosylation; cutis laxa.
Copyright © 2017 American Society of Human Genetics. All rights reserved.
Figures
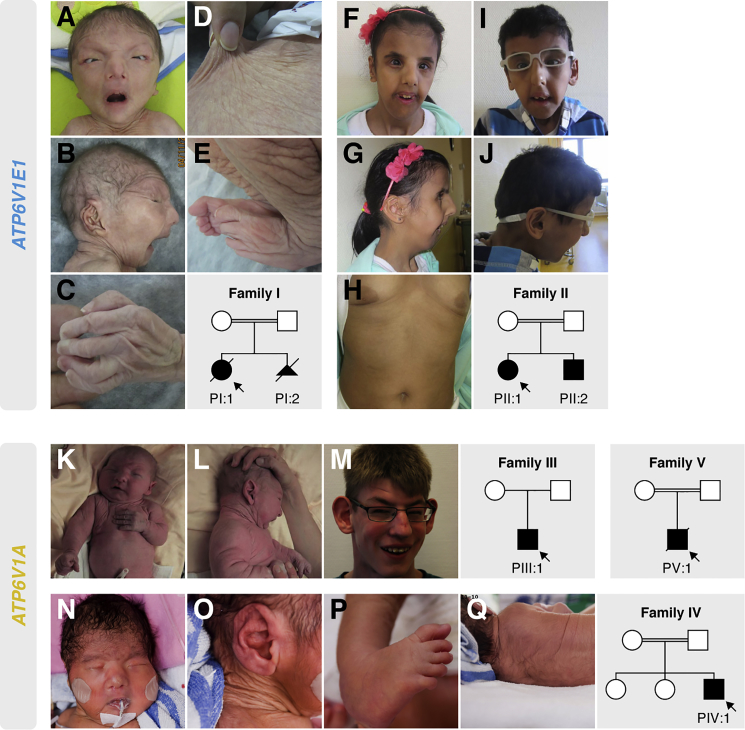
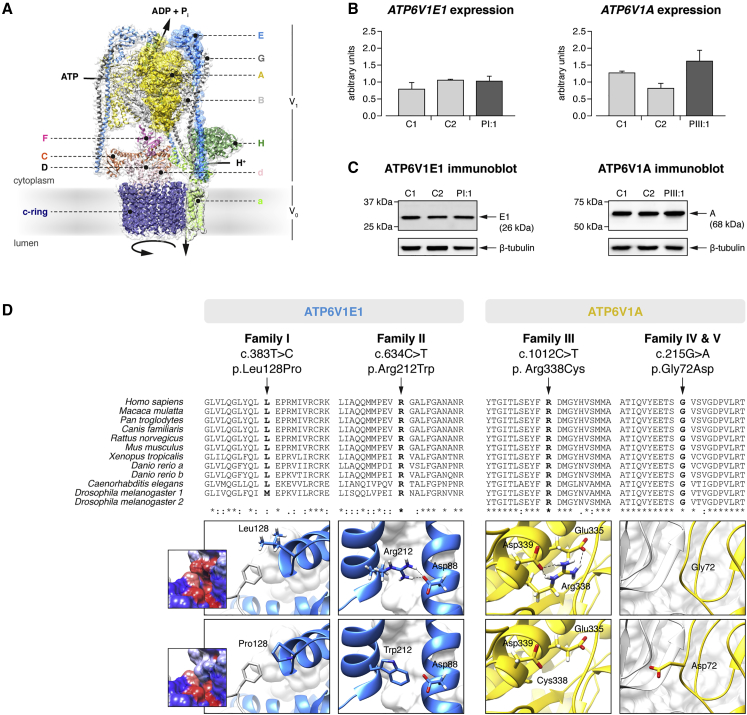
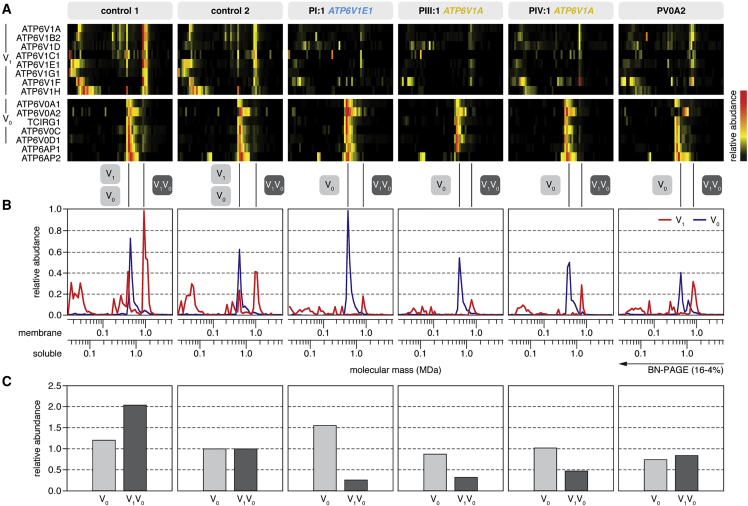
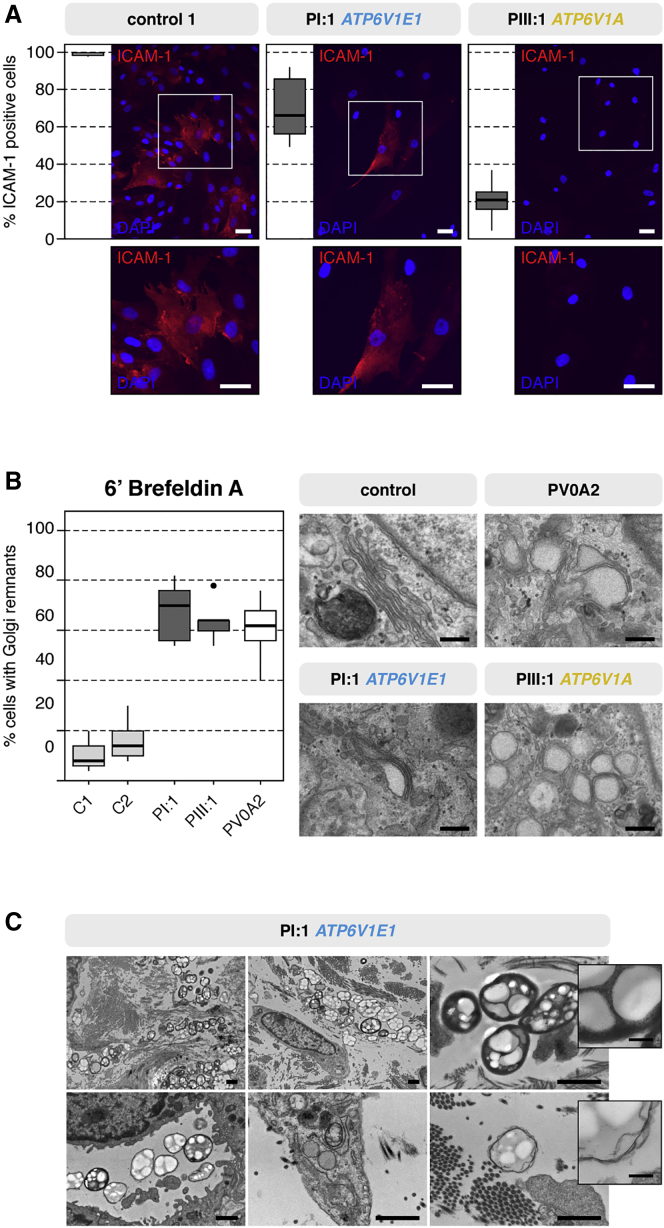
Comment in
-
[Autosomal recessive cutis laxa: New genes identified].Ann Dermatol Venereol. 2018 Aug-Sep;145(8-9):554-555. doi: 10.1016/j.annder.2018.05.003. Epub 2018 Jul 26. Ann Dermatol Venereol. 2018. PMID: 30056993 French. No abstract available.
References
-
- Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007;8:917–929. - PubMed
-
- Guillard M., Dimopoulou A., Fischer B., Morava E., Lefeber D.J., Kornak U., Wevers R.A. Vacuolar H+-ATPase meets glycosylation in patients with cutis laxa. Biochim. Biophys. Acta. 2009;1792:903–914. - PubMed
-
- Karet F.E., Finberg K.E., Nelson R.D., Nayir A., Mocan H., Sanjad S.A., Rodriguez-Soriano J., Santos F., Cremers C.W.R.J., Di Pietro A. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat. Genet. 1999;21:84–90. - PubMed
-
- Kortüm F., Caputo V., Bauer C.K., Stella L., Ciolfi A., Alawi M., Bocchinfuso G., Flex E., Paolacci S., Dentici M.L. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat. Genet. 2015;47:661–667. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
