

From Genomes to Phenotypes: Traitar, the Microbial Trait Analyzer
- PMID: 28066816
- PMCID: PMC5192078
- DOI: 10.1128/mSystems.00101-16
From Genomes to Phenotypes: Traitar, the Microbial Trait Analyzer
Abstract
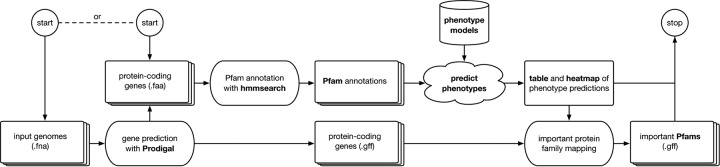
The number of sequenced genomes is growing exponentially, profoundly shifting the bottleneck from data generation to genome interpretation. Traits are often used to characterize and distinguish bacteria and are likely a driving factor in microbial community composition, yet little is known about the traits of most microbes. We describe Traitar, the microbial trait analyzer, which is a fully automated software package for deriving phenotypes from a genome sequence. Traitar provides phenotype classifiers to predict 67 traits related to the use of various substrates as carbon and energy sources, oxygen requirement, morphology, antibiotic susceptibility, proteolysis, and enzymatic activities. Furthermore, it suggests protein families associated with the presence of particular phenotypes. Our method uses L1-regularized L2-loss support vector machines for phenotype assignments based on phyletic patterns of protein families and their evolutionary histories across a diverse set of microbial species. We demonstrate reliable phenotype assignment for Traitar to bacterial genomes from 572 species of eight phyla, also based on incomplete single-cell genomes and simulated draft genomes. We also showcase its application in metagenomics by verifying and complementing a manual metabolic reconstruction of two novel Clostridiales species based on draft genomes recovered from commercial biogas reactors. Traitar is available at https://github.com/hzi-bifo/traitar. IMPORTANCE Bacteria are ubiquitous in our ecosystem and have a major impact on human health, e.g., by supporting digestion in the human gut. Bacterial communities can also aid in biotechnological processes such as wastewater treatment or decontamination of polluted soils. Diverse bacteria contribute with their unique capabilities to the functioning of such ecosystems, but lab experiments to investigate those capabilities are labor-intensive. Major advances in sequencing techniques open up the opportunity to study bacteria by their genome sequences. For this purpose, we have developed Traitar, software that predicts traits of bacteria on the basis of their genomes. It is applicable to studies with tens or hundreds of bacterial genomes. Traitar may help researchers in microbiology to pinpoint the traits of interest, reducing the amount of wet lab work required.
Keywords: ancestral trait reconstruction; genotype-phenotype inference; metagenomics; microbial traits; phenotypes; phyletic patterns; single-cell genomics; support vector machines.
Figures






References
-
- Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K-i, Ludwig W, Whitman WB. 2012. Bergey’s manual of systematic bacteriology. Springer, New York, NY.
-
- Bai Y, Müller DB, Srinivas G, Garrido-Oter R, Potthoff E, Rott M, Dombrowski N, Münch PC, Spaepen S, Remus-Emsermann M, Hüttel B, McHardy AC, Vorholt JA, Schulze-Lefert P. 2015. Functional overlap of the Arabidopsis leaf and root microbiota. Nature 528:364–369. doi: 10.1038/nature16192. - DOI - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
