

Quantum-chemical insights from deep tensor neural networks
- PMID: 28067221
- PMCID: PMC5228054
- DOI: 10.1038/ncomms13890
Quantum-chemical insights from deep tensor neural networks
Abstract
Learning from data has led to paradigm shifts in a multitude of disciplines, including web, text and image search, speech recognition, as well as bioinformatics. Can machine learning enable similar breakthroughs in understanding quantum many-body systems? Here we develop an efficient deep learning approach that enables spatially and chemically resolved insights into quantum-mechanical observables of molecular systems. We unify concepts from many-body Hamiltonians with purpose-designed deep tensor neural networks, which leads to size-extensive and uniformly accurate (1 kcal mol-1) predictions in compositional and configurational chemical space for molecules of intermediate size. As an example of chemical relevance, the model reveals a classification of aromatic rings with respect to their stability. Further applications of our model for predicting atomic energies and local chemical potentials in molecules, reliable isomer energies, and molecules with peculiar electronic structure demonstrate the potential of machine learning for revealing insights into complex quantum-chemical systems.
Figures

 , which is repeatedly refined by interactions vij. The interactions depend on the current representation
, which is repeatedly refined by interactions vij. The interactions depend on the current representation  , as well as the distance Dij to an atom j. After T iterations, an energy contribution Ei is predicted for the final coefficient vector
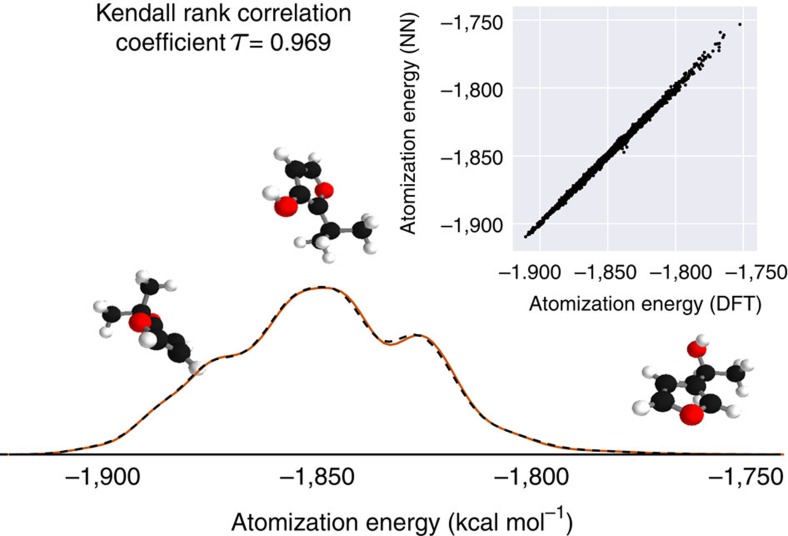
, as well as the distance Dij to an atom j. After T iterations, an energy contribution Ei is predicted for the final coefficient vector  . The molecular energy E is the sum over these atomic contributions. (c) Mean absolute errors of predictions for the GDB-9 dataset of 133,885 molecules as a function of the number of atoms. The employed neural network uses two interaction passes (T=2) and 50,000 reference calculation during training. The inset shows the error of an equivalent network trained on 5,000 GDB-9 molecules with 20 or more atoms, as small molecules with 15 or less atoms are added to the training set. (d) Extract from the calculated (black) and predicted (orange) molecular dynamics trajectory of toluene. The curve on the right shows the agreement of the predicted and calculated energy distributions. (e) Energy contribution Eprobe (or local chemical potential
. The molecular energy E is the sum over these atomic contributions. (c) Mean absolute errors of predictions for the GDB-9 dataset of 133,885 molecules as a function of the number of atoms. The employed neural network uses two interaction passes (T=2) and 50,000 reference calculation during training. The inset shows the error of an equivalent network trained on 5,000 GDB-9 molecules with 20 or more atoms, as small molecules with 15 or less atoms are added to the training set. (d) Extract from the calculated (black) and predicted (orange) molecular dynamics trajectory of toluene. The curve on the right shows the agreement of the predicted and calculated energy distributions. (e) Energy contribution Eprobe (or local chemical potential  , see text) of a hydrogen test charge on a
, see text) of a hydrogen test charge on a  isosurface for various molecules from the GDB-9 dataset for a DTNN model with T=2.
isosurface for various molecules from the GDB-9 dataset for a DTNN model with T=2.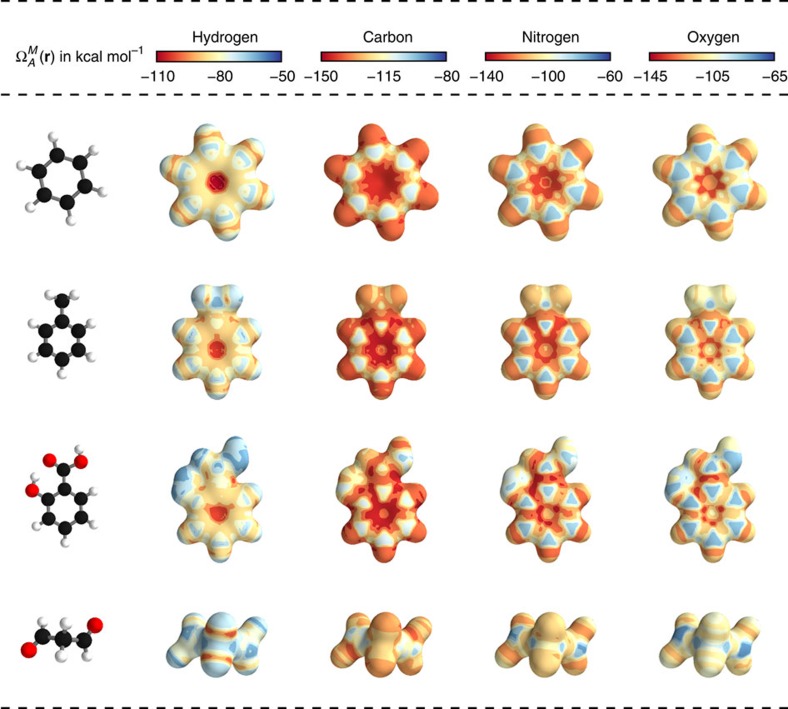
 =3.8 Å−2 (the index i is used to sum over all atoms of the corresponding molecule). The molecules shown are (in order from top to bottom of the figure): benzene, toluene, salicylic acid and malondehyde. Atom colouring: carbon=black, hydrogen=white, oxygen=red.
=3.8 Å−2 (the index i is used to sum over all atoms of the corresponding molecule). The molecules shown are (in order from top to bottom of the figure): benzene, toluene, salicylic acid and malondehyde. Atom colouring: carbon=black, hydrogen=white, oxygen=red.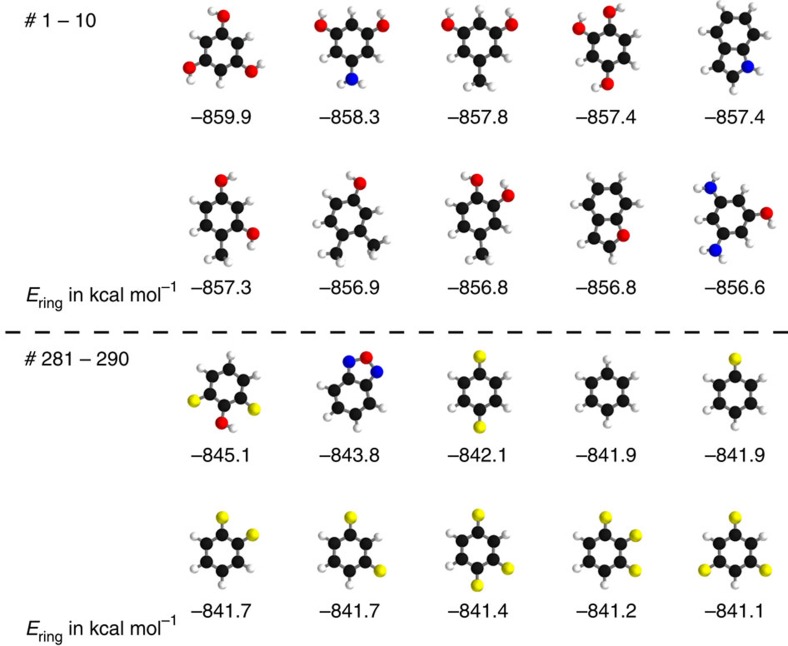
References
-
- Kang B. & Ceder G. Battery materials for ultrafast charging and discharging. Nature 458, 190–193 (2009). - PubMed
-
- Nørskov J. K., Bligaard T., Rossmeisl J. & Christensen C. H. Towards the computational design of solid catalysts. Nat. Chem. 1, 37–46 (2009). - PubMed
-
- Hachmann J. et al.. The Harvard clean energy project: large-scale computational screening and design of organic photo-voltaics on the world community grid. J. Phys. Chem. Lett. 2, 2241–2251 (2011).
-
- Pyzer-Knapp E. O., Suh C., Gomez-Bombarelli R., Aguilera-Iparraguirre J. & Aspuru-Guzik A. What is high-throughput virtual screening? A perspective from organic materials discovery. Annu. Rev. Mater. Res. 45, 195–216 (2015).
-
- Curtarolo S. et al.. The high-throughput highway to computational materials design. Nat. Mater. 12, 191–201 (2013). - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
