

Targeting deregulated AMPK/mTORC1 pathways improves muscle function in myotonic dystrophy type I
- PMID: 28067669
- PMCID: PMC5272183
- DOI: 10.1172/JCI89616
Targeting deregulated AMPK/mTORC1 pathways improves muscle function in myotonic dystrophy type I
Abstract
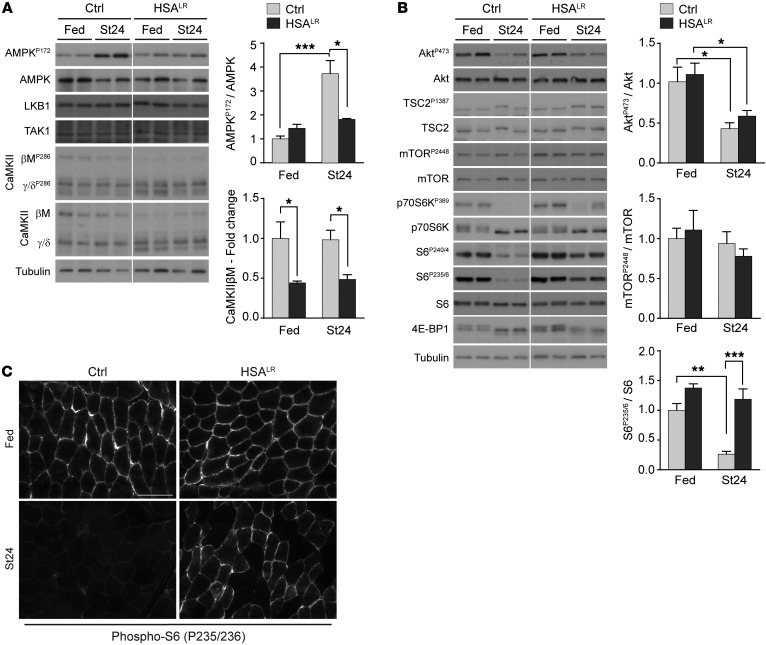
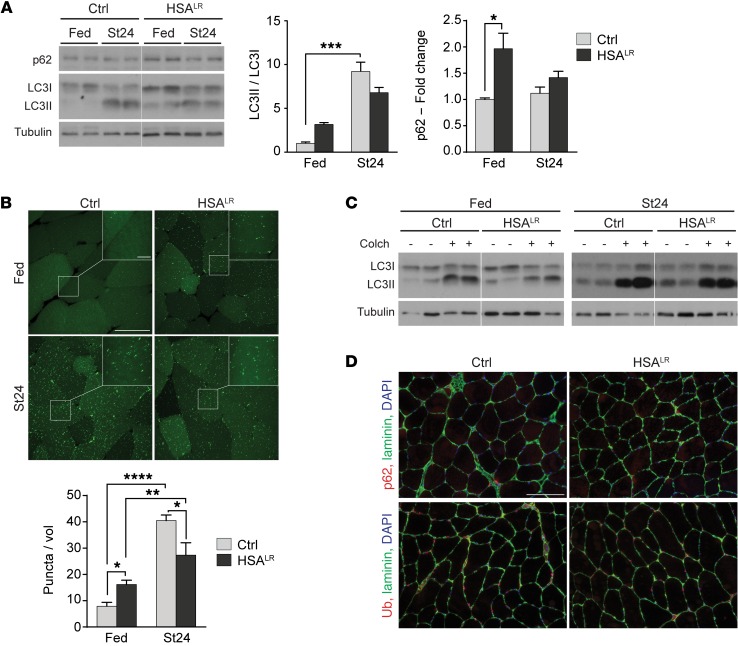
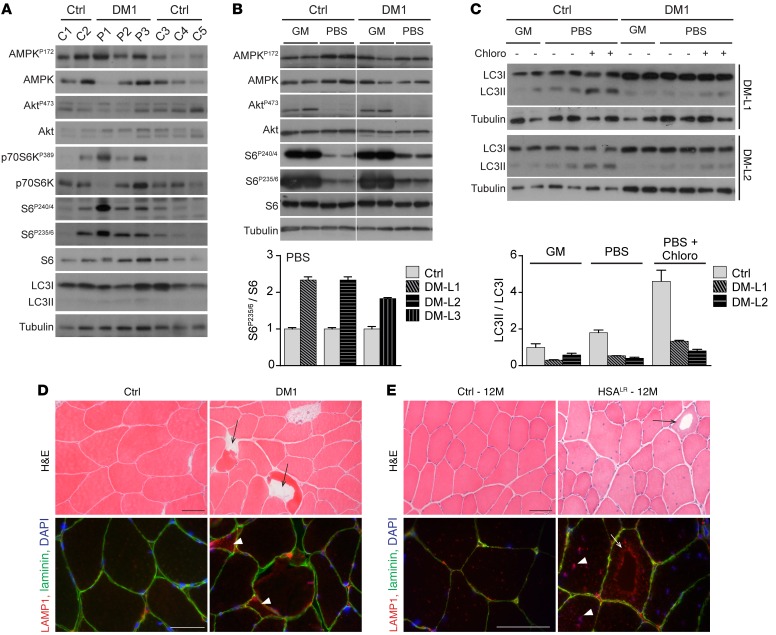
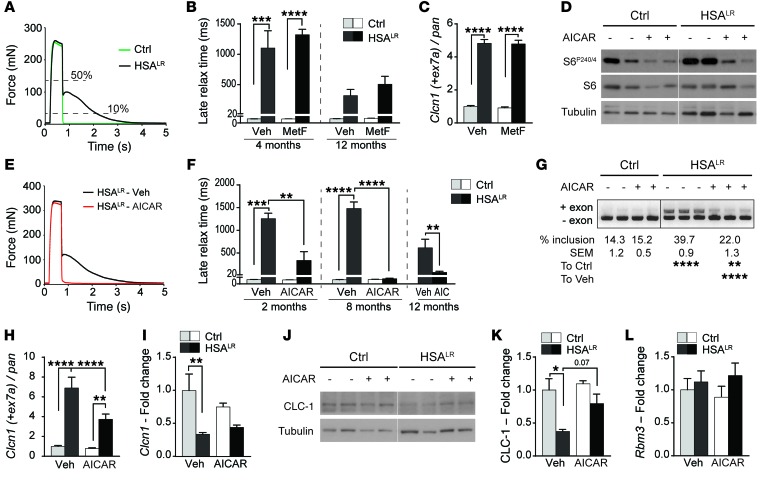
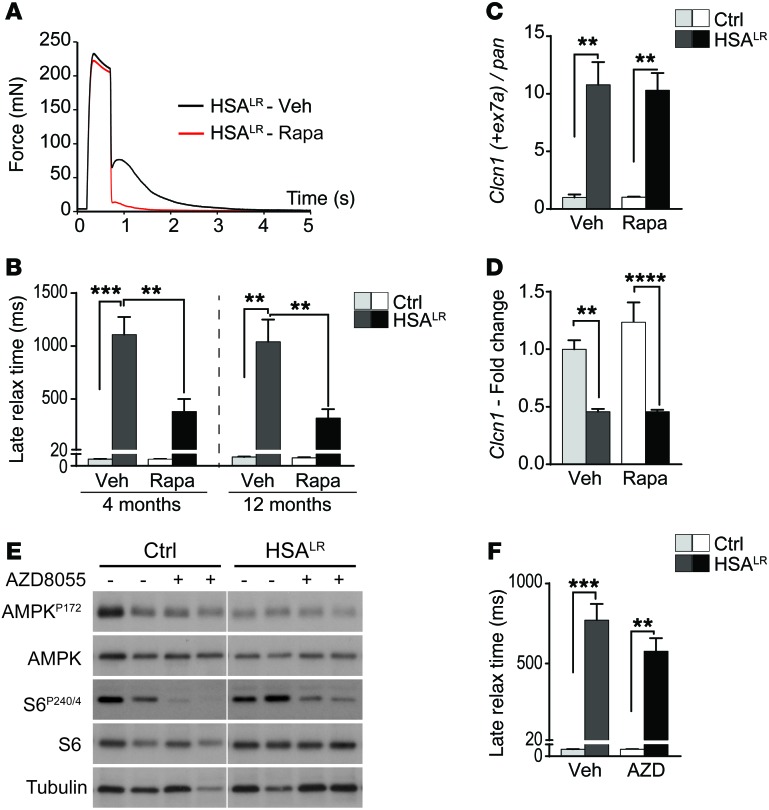
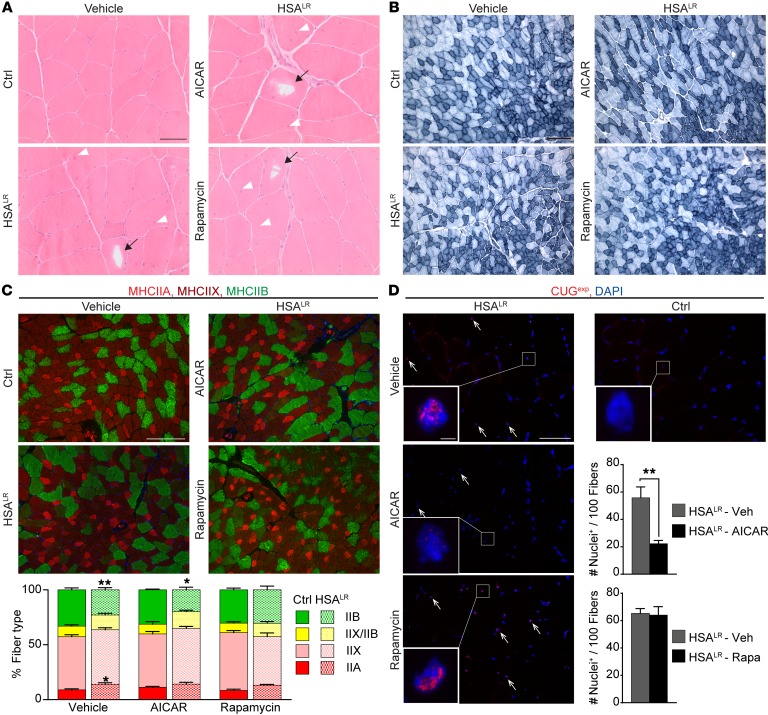
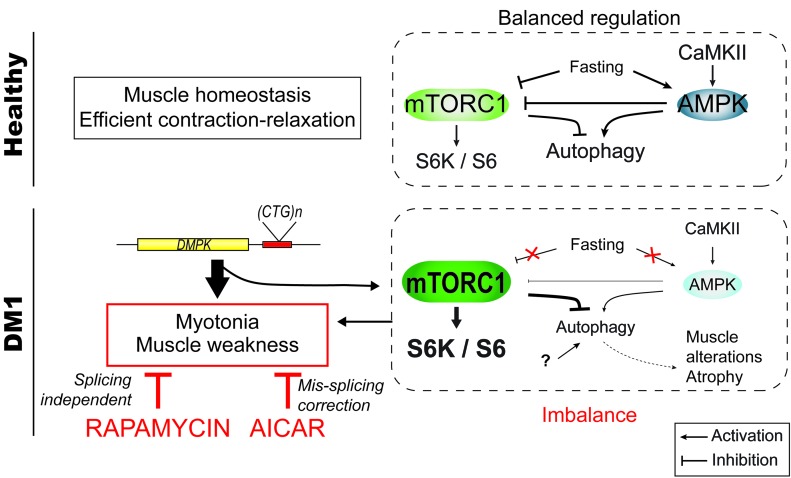
Myotonic dystrophy type I (DM1) is a disabling multisystemic disease that predominantly affects skeletal muscle. It is caused by expanded CTG repeats in the 3'-UTR of the dystrophia myotonica protein kinase (DMPK) gene. RNA hairpins formed by elongated DMPK transcripts sequester RNA-binding proteins, leading to mis-splicing of numerous pre-mRNAs. Here, we have investigated whether DM1-associated muscle pathology is related to deregulation of central metabolic pathways, which may identify potential therapeutic targets for the disease. In a well-characterized mouse model for DM1 (HSALR mice), activation of AMPK signaling in muscle was impaired under starved conditions, while mTORC1 signaling remained active. In parallel, autophagic flux was perturbed in HSALR muscle and in cultured human DM1 myotubes. Pharmacological approaches targeting AMPK/mTORC1 signaling greatly ameliorated muscle function in HSALR mice. AICAR, an AMPK activator, led to a strong reduction of myotonia, which was accompanied by partial correction of misregulated alternative splicing. Rapamycin, an mTORC1 inhibitor, improved muscle relaxation and increased muscle force in HSALR mice without affecting splicing. These findings highlight the involvement of AMPK/mTORC1 deregulation in DM1 muscle pathophysiology and may open potential avenues for the treatment of this disease.
Conflict of interest statement
M. Sinnreich owns shares of Novartis and is coinventor on a patent application for drug discovery in DM1 (EP 16/166212.7). M. Sinnreich’s institution (University Hospital Basel) has received research support from CSL Behring and Roche, not in relation to this study. C. Angelini is part of the European Board of Genzyme-Sanofi.
Figures


Similar articles
-
Pharmacological and physiological activation of AMPK improves the spliceopathy in DM1 mouse muscles.Hum Mol Genet. 2018 Oct 1;27(19):3361-3376. doi: 10.1093/hmg/ddy245. Hum Mol Genet. 2018. PMID: 29982462 Free PMC article.
-
The AMPK allosteric activator MK-8722 improves the histology and spliceopathy in myotonic dystrophy type 1 (DM1) skeletal muscle.FASEB J. 2024 Dec 15;38(23):e70199. doi: 10.1096/fj.202401145RR. FASEB J. 2024. PMID: 39611312
-
Antagonistic control of muscle cell size by AMPK and mTORC1.Cell Cycle. 2011 Aug 15;10(16):2640-6. doi: 10.4161/cc.10.16.17102. Epub 2011 Aug 15. Cell Cycle. 2011. PMID: 21799304
-
Correction of RNA-Binding Protein CUGBP1 and GSK3β Signaling as Therapeutic Approach for Congenital and Adult Myotonic Dystrophy Type 1.Int J Mol Sci. 2019 Dec 21;21(1):94. doi: 10.3390/ijms21010094. Int J Mol Sci. 2019. PMID: 31877772 Free PMC article. Review.
-
Molecular Therapies for Myotonic Dystrophy Type 1: From Small Drugs to Gene Editing.Int J Mol Sci. 2022 Apr 21;23(9):4622. doi: 10.3390/ijms23094622. Int J Mol Sci. 2022. PMID: 35563013 Free PMC article. Review.
Cited by
-
Myotonic Dystrophies: A Genetic Overview.Genes (Basel). 2022 Feb 17;13(2):367. doi: 10.3390/genes13020367. Genes (Basel). 2022. PMID: 35205411 Free PMC article. Review.
-
Pharmacological and physiological activation of AMPK improves the spliceopathy in DM1 mouse muscles.Hum Mol Genet. 2018 Oct 1;27(19):3361-3376. doi: 10.1093/hmg/ddy245. Hum Mol Genet. 2018. PMID: 29982462 Free PMC article.
-
Control of satellite cell function in muscle regeneration and its disruption in ageing.Nat Rev Mol Cell Biol. 2022 Mar;23(3):204-226. doi: 10.1038/s41580-021-00421-2. Epub 2021 Oct 18. Nat Rev Mol Cell Biol. 2022. PMID: 34663964 Review.
-
Mechanisms of skeletal muscle wasting in a mouse model for myotonic dystrophy type 1.Hum Mol Genet. 2018 Aug 15;27(16):2789-2804. doi: 10.1093/hmg/ddy192. Hum Mol Genet. 2018. PMID: 29771332 Free PMC article.
-
Alternative splicing dysregulation across tissue and therapeutic approaches in a mouse model of myotonic dystrophy type 1.Mol Ther Nucleic Acids. 2024 Sep 13;35(4):102338. doi: 10.1016/j.omtn.2024.102338. eCollection 2024 Dec 10. Mol Ther Nucleic Acids. 2024. PMID: 39391766 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
