

Modulation of Intersectin-1s Lung Expression Induces Obliterative Remodeling and Severe Plexiform Arteriopathy in the Murine Pulmonary Vascular Bed
- PMID: 28068512
- PMCID: PMC5389368
- DOI: 10.1016/j.ajpath.2016.11.012
Modulation of Intersectin-1s Lung Expression Induces Obliterative Remodeling and Severe Plexiform Arteriopathy in the Murine Pulmonary Vascular Bed
Abstract
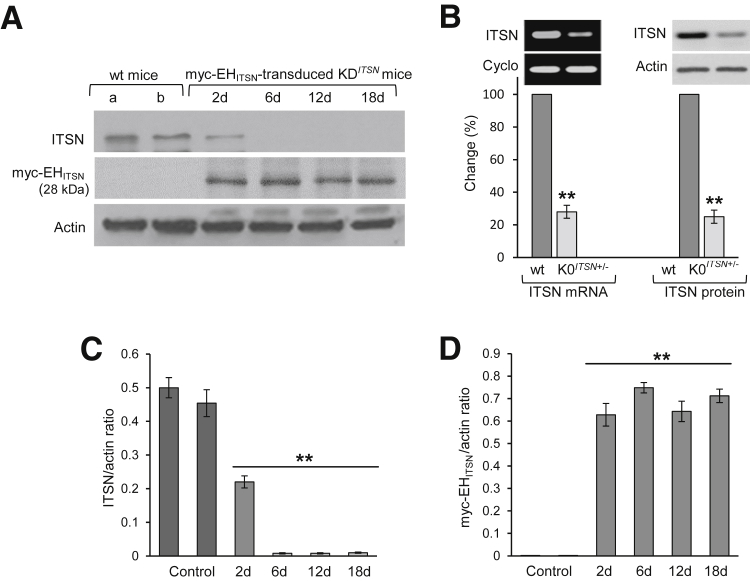
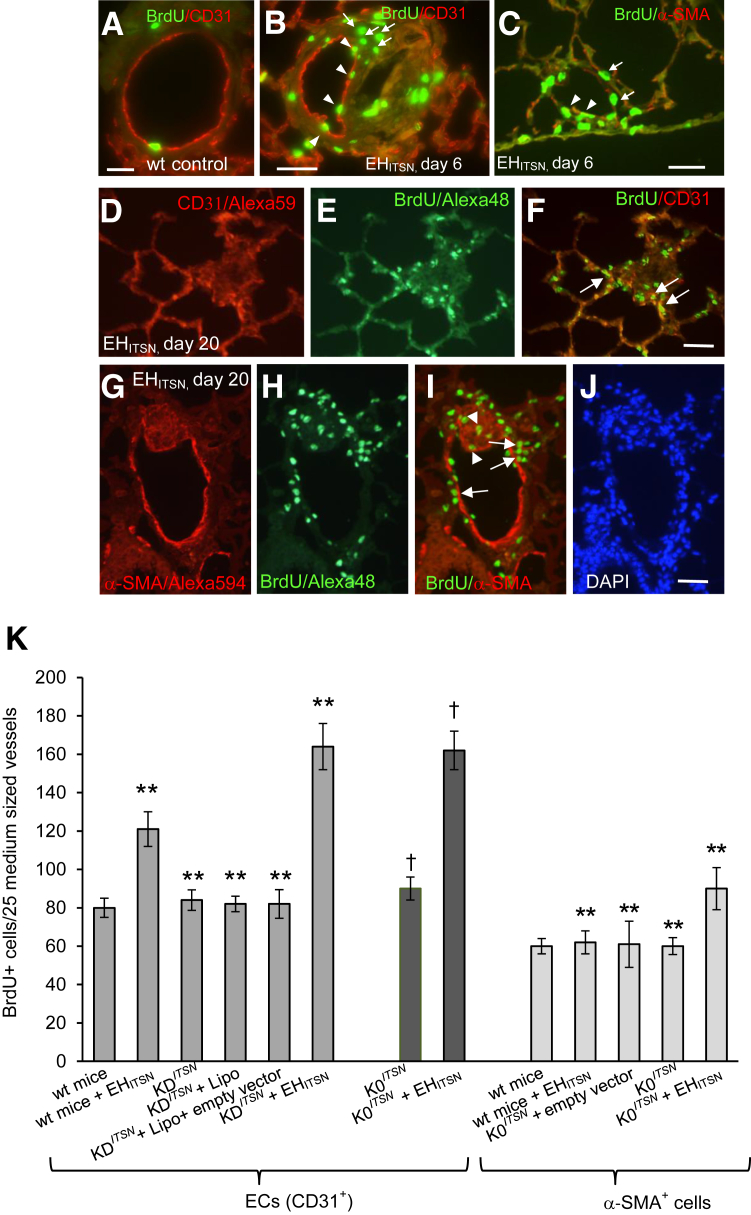
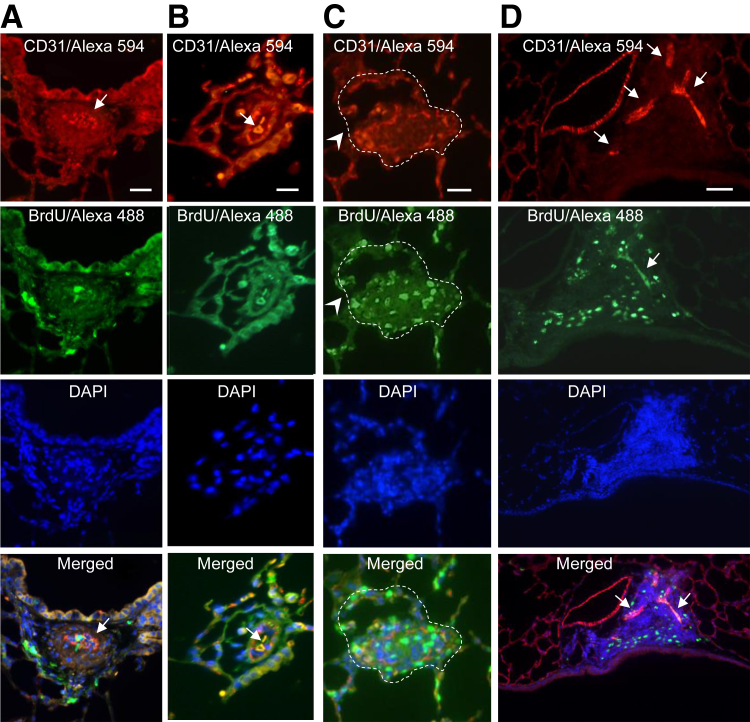
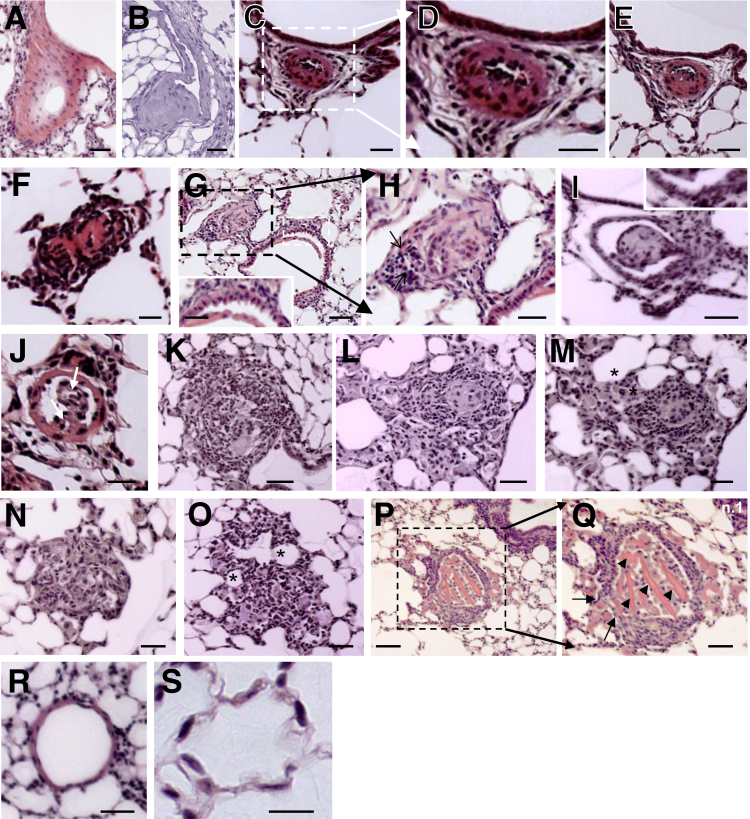
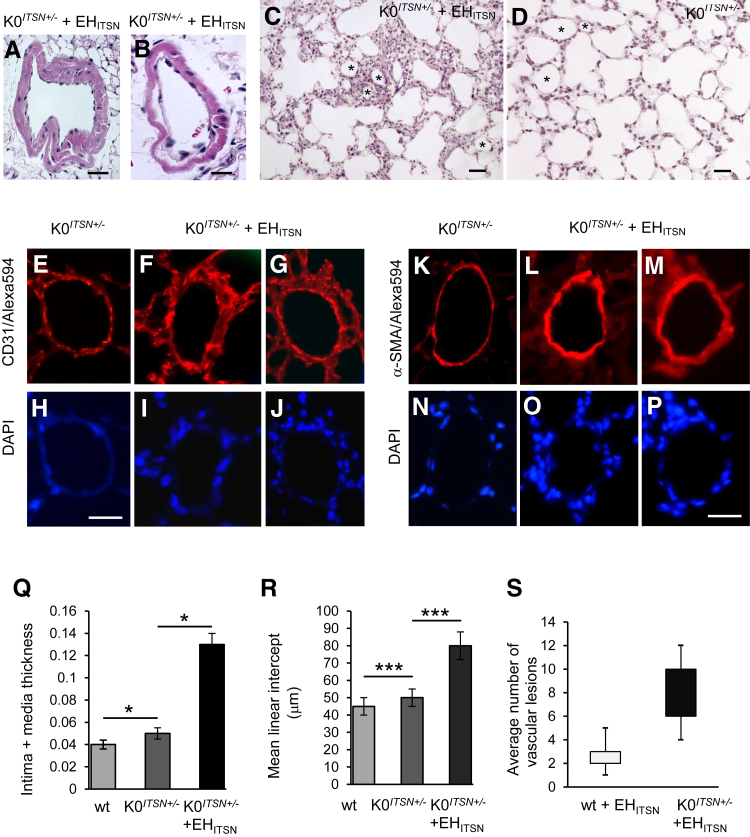
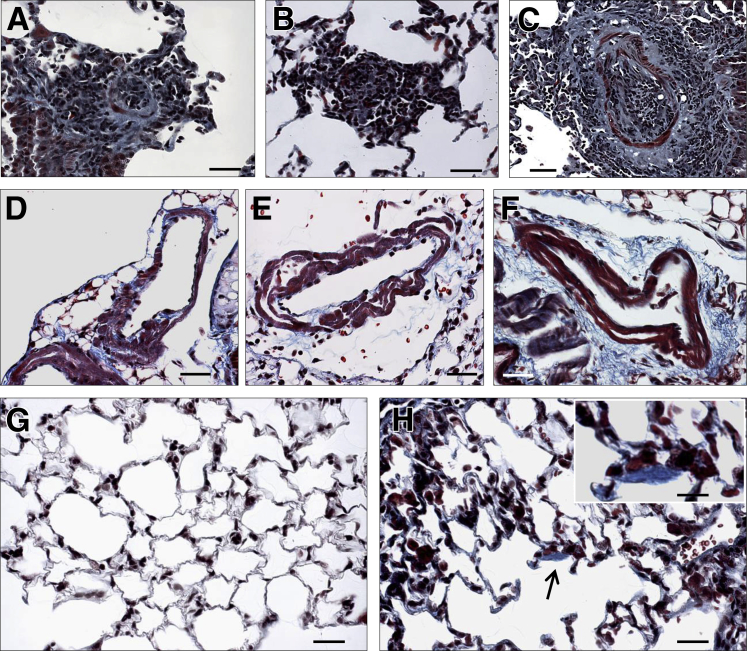
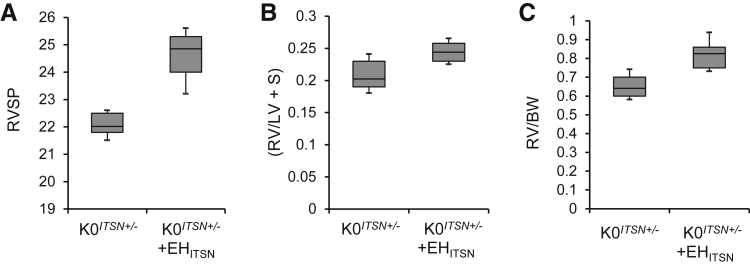
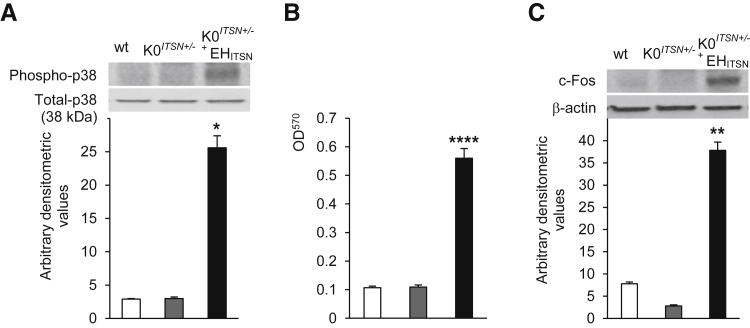
Murine models of pulmonary arterial hypertension (PAH) that recapitulate the plexiform and obliterative arteriopathy seen in PAH patients and help in defining the molecular mechanisms involved are missing. Herein, we investigated whether intersectin-1s (ITSN) deficiency and prolonged lung expression of an ITSN fragment with endothelial cell (EC) proliferative potential (EHITSN), present in the lungs of PAH animal models and human patients, induce formation of plexiform/obliterative lesions and defined the molecular mechanisms involved. ITSN-deficient mice (knockout/heterozygous and knockdown) were subjected to targeted lung delivery of EHITSN via liposomes for 20 days. Immunohistochemistry and histological and morphometric analyses revealed a twofold increase in proliferative ECs and a 1.35-fold increase in proliferative α-smooth muscle actin-positive cells in the lungs of ITSN-deficient mice, transduced with the EHITSN relative to wild-type littermates. Treated mice developed severe medial wall hypertrophy, intima proliferation, and various forms of obliterative and plexiform-like lesions in pulmonary arteries, similar to PAH patients. Hemodynamic measurements indicated modest increases in the right ventricular systolic pressure and right ventricle hypertrophy. Transcriptional and protein assays of lung tissue indicated p38MAPK-dependent activation of Elk-1 transcription factor and increased expression of c-Fos gene. This unique murine model of PAH-like plexiform/obliterative arteriopathy induced via a two-hit pathophysiological mechanism without hypoxia provides novel druggable targets to ameliorate and, perhaps, reverse the EC plexiform phenotype in severe human PAH.
Copyright © 2017 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Epsin15 Homology Domains: Role in the Pathogenesis of Pulmonary Arterial Hypertension.Front Physiol. 2018 Oct 2;9:1393. doi: 10.3389/fphys.2018.01393. eCollection 2018. Front Physiol. 2018. PMID: 30333761 Free PMC article.
-
A novel p38 mitogen-activated protein kinase/Elk-1 transcription factor-dependent molecular mechanism underlying abnormal endothelial cell proliferation in plexogenic pulmonary arterial hypertension.J Biol Chem. 2013 Sep 6;288(36):25701-25716. doi: 10.1074/jbc.M113.502674. Epub 2013 Jul 26. J Biol Chem. 2013. PMID: 23893408 Free PMC article.
-
Intersectin-1s deficiency in pulmonary pathogenesis.Respir Res. 2017 Sep 6;18(1):168. doi: 10.1186/s12931-017-0652-4. Respir Res. 2017. PMID: 28874189 Free PMC article. Review.
-
Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans Through Hypoxia-Inducible Factor-2α.Circulation. 2016 Jun 14;133(24):2447-58. doi: 10.1161/CIRCULATIONAHA.116.021494. Epub 2016 Apr 25. Circulation. 2016. PMID: 27143681 Free PMC article.
-
Plexiform Arteriopathy in Rodent Models of Pulmonary Arterial Hypertension.Am J Pathol. 2019 Jun;189(6):1133-1144. doi: 10.1016/j.ajpath.2019.02.005. Epub 2019 Mar 26. Am J Pathol. 2019. PMID: 30926336 Free PMC article. Review.
Cited by
-
Epsin15 Homology Domains: Role in the Pathogenesis of Pulmonary Arterial Hypertension.Front Physiol. 2018 Oct 2;9:1393. doi: 10.3389/fphys.2018.01393. eCollection 2018. Front Physiol. 2018. PMID: 30333761 Free PMC article.
-
Up-Regulation of the Long Noncoding RNA X-Inactive-Specific Transcript and the Sex Bias in Pulmonary Arterial Hypertension.Am J Pathol. 2021 Jun;191(6):1135-1150. doi: 10.1016/j.ajpath.2021.03.009. Epub 2021 Apr 6. Am J Pathol. 2021. PMID: 33836164 Free PMC article.
-
The Impact of Sex Chromosomes in the Sexual Dimorphism of Pulmonary Arterial Hypertension.Am J Pathol. 2022 Apr;192(4):582-594. doi: 10.1016/j.ajpath.2022.01.005. Epub 2022 Feb 1. Am J Pathol. 2022. PMID: 35114193 Free PMC article. Review.
-
Alk5/Runx1 signaling mediated by extracellular vesicles promotes vascular repair in acute respiratory distress syndrome.Clin Transl Med. 2018 Jun 22;7(1):19. doi: 10.1186/s40169-018-0197-2. Clin Transl Med. 2018. PMID: 29931538 Free PMC article.
-
Dysregulation of the Long Noncoding RNA X-Inactive-Specific Transcript Expression in Male Patients with Pulmonary Arterial Hypertension.Am J Pathol. 2024 Aug;194(8):1592-1606. doi: 10.1016/j.ajpath.2024.04.005. Epub 2024 May 3. Am J Pathol. 2024. PMID: 38705381 Free PMC article.
References
-
- Tuder R.M., Cool C.D., Yeager M., Taraseviciene-Stewart L., Bull T.M., Voelkel N.F. The pathobiology of pulmonary hypertension: endothelium. Clin Chest Med. 2001;22:405–418. - PubMed
-
- Voelkel N.F., Cool C. Pathology of pulmonary hypertension. Cardiol Clin. 2004;22:343–351. v. - PubMed
-
- Tuder R.M., Abman S.H., Braun T., Capron F., Stevens T., Thistlethwaite P.A., Haworth S.G. Development and pathology of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S3–S9. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
