

BCL9L Dysfunction Impairs Caspase-2 Expression Permitting Aneuploidy Tolerance in Colorectal Cancer
- PMID: 28073006
- PMCID: PMC5225404
- DOI: 10.1016/j.ccell.2016.11.001
BCL9L Dysfunction Impairs Caspase-2 Expression Permitting Aneuploidy Tolerance in Colorectal Cancer
Abstract
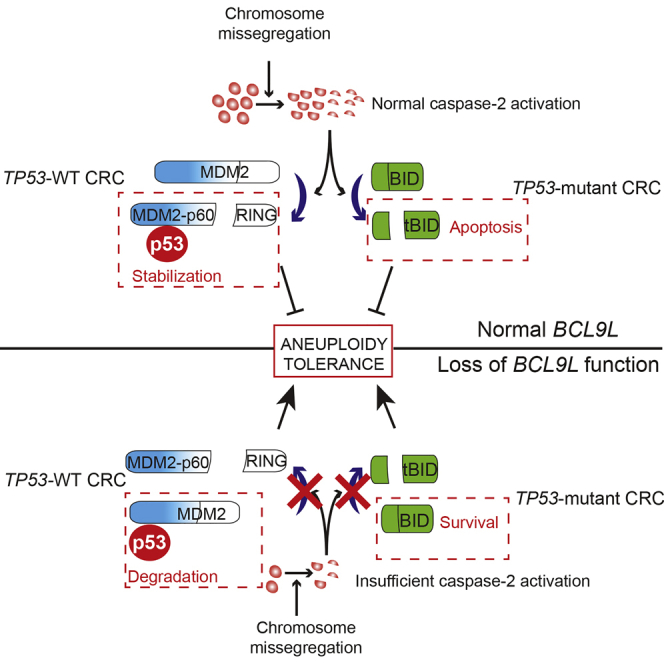
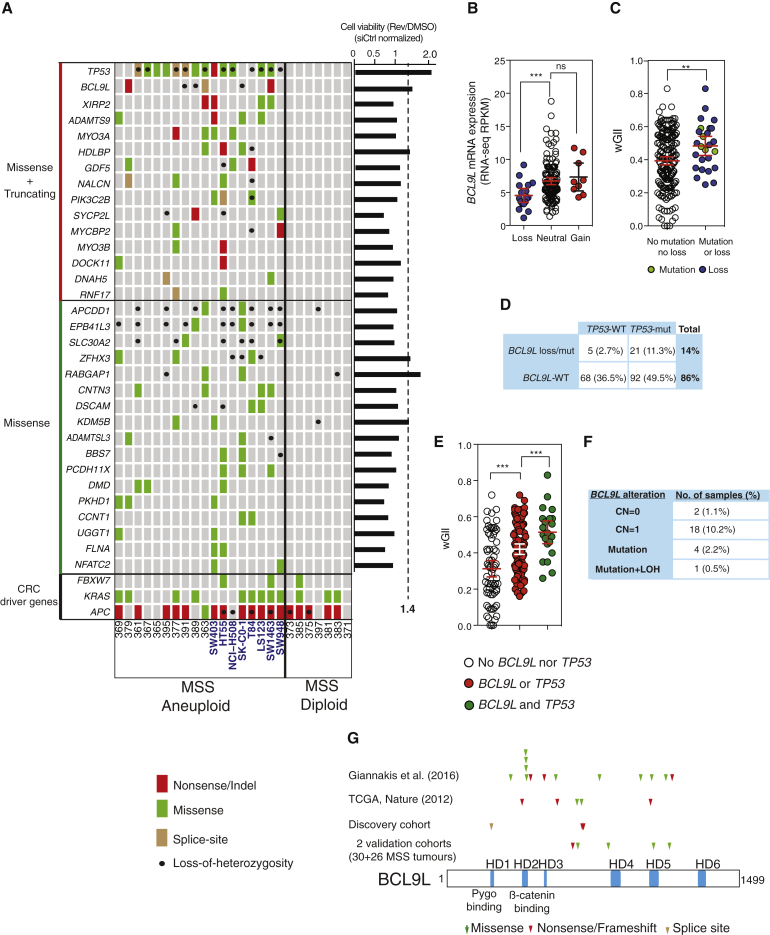
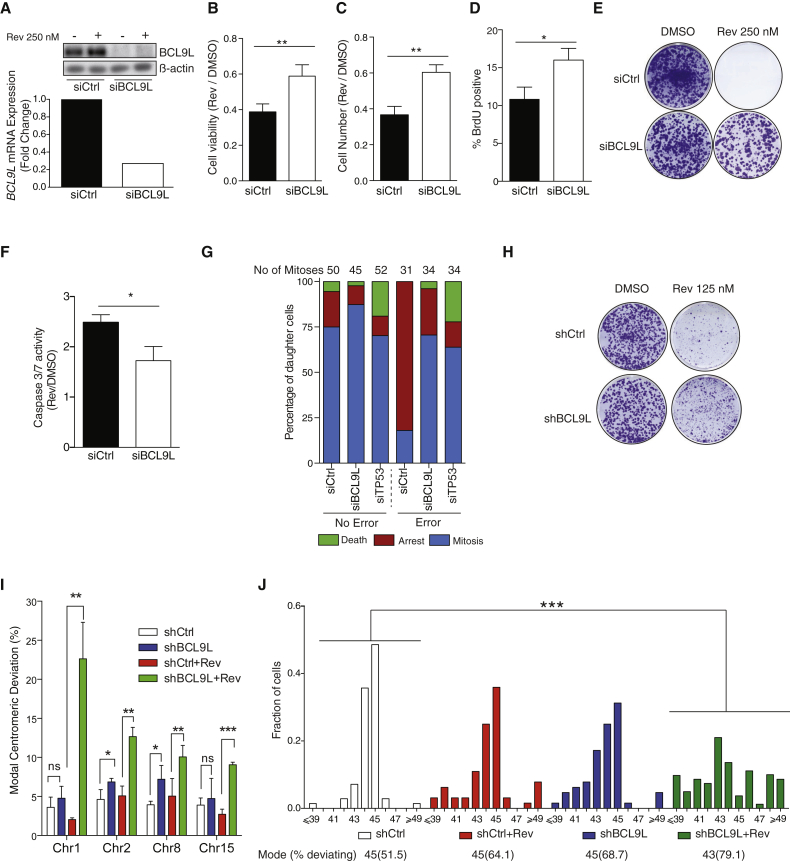
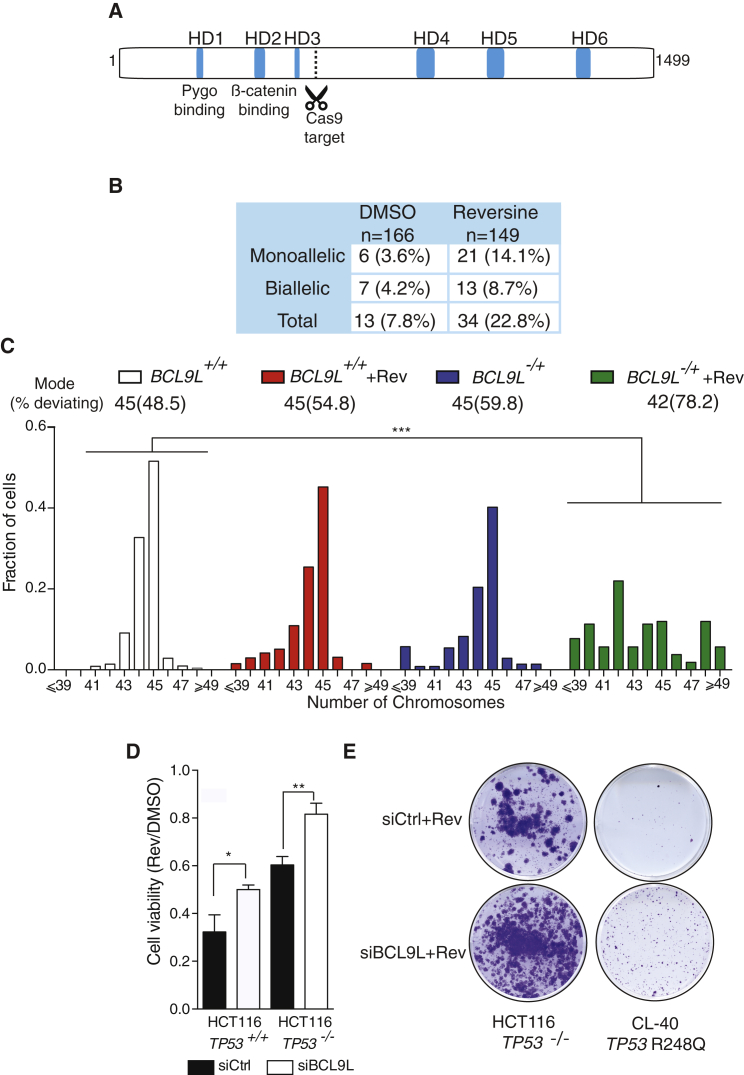
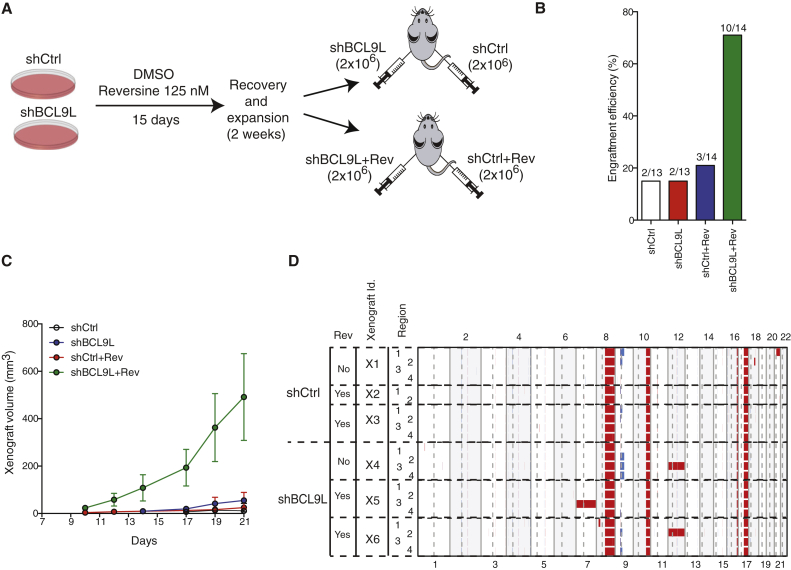
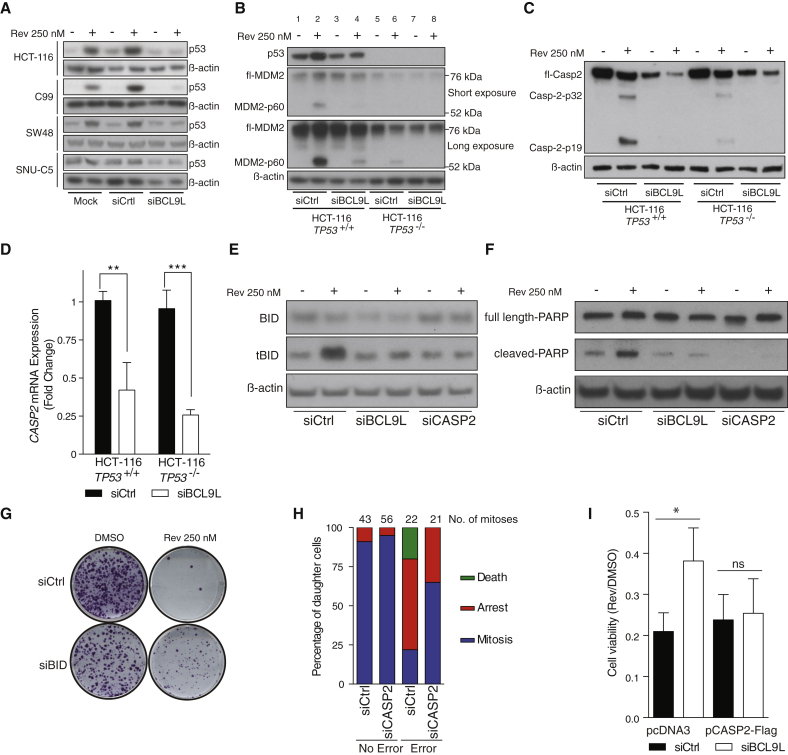
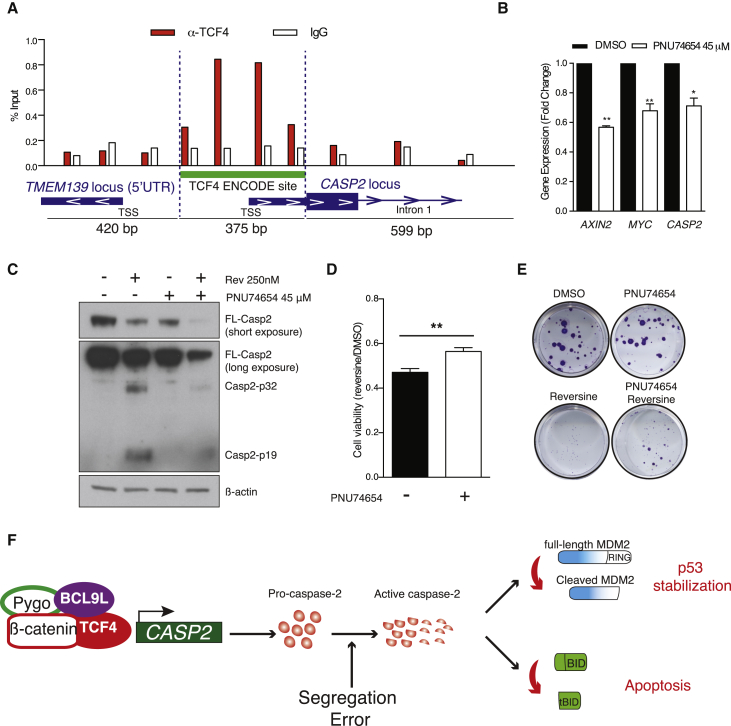
Chromosomal instability (CIN) contributes to cancer evolution, intratumor heterogeneity, and drug resistance. CIN is driven by chromosome segregation errors and a tolerance phenotype that permits the propagation of aneuploid genomes. Through genomic analysis of colorectal cancers and cell lines, we find frequent loss of heterozygosity and mutations in BCL9L in aneuploid tumors. BCL9L deficiency promoted tolerance of chromosome missegregation events, propagation of aneuploidy, and genetic heterogeneity in xenograft models likely through modulation of Wnt signaling. We find that BCL9L dysfunction contributes to aneuploidy tolerance in both TP53-WT and mutant cells by reducing basal caspase-2 levels and preventing cleavage of MDM2 and BID. Efforts to exploit aneuploidy tolerance mechanisms and the BCL9L/caspase-2/BID axis may limit cancer diversity and evolution.
Keywords: BCL9L; BID; aneuploidy tolerance; caspase-2; chromosomal instability; chromosome segregation errors; colorectal cancer evolution; intratumor heterogeneity; mitotic checkpoint; p53.
Copyright © 2017 The Francis Crick Institute. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Albuquerque C., Breukel C., van del Luijt R., Fidalgo P., Lage P., Slors F.J.M., Nobre-Leitao C., Fodde R., Smits R. The “just-right” signaling model: APC somatic mutations are selected based on a specific level of activation of the β-catenin signaling cascade. Hum. Mol. Genet. 2002;11:1549–1560. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
