

Histones and Neutrophil Extracellular Traps Enhance Tubular Necrosis and Remote Organ Injury in Ischemic AKI
- PMID: 28073931
- PMCID: PMC5461800
- DOI: 10.1681/ASN.2016080925
Histones and Neutrophil Extracellular Traps Enhance Tubular Necrosis and Remote Organ Injury in Ischemic AKI
Abstract
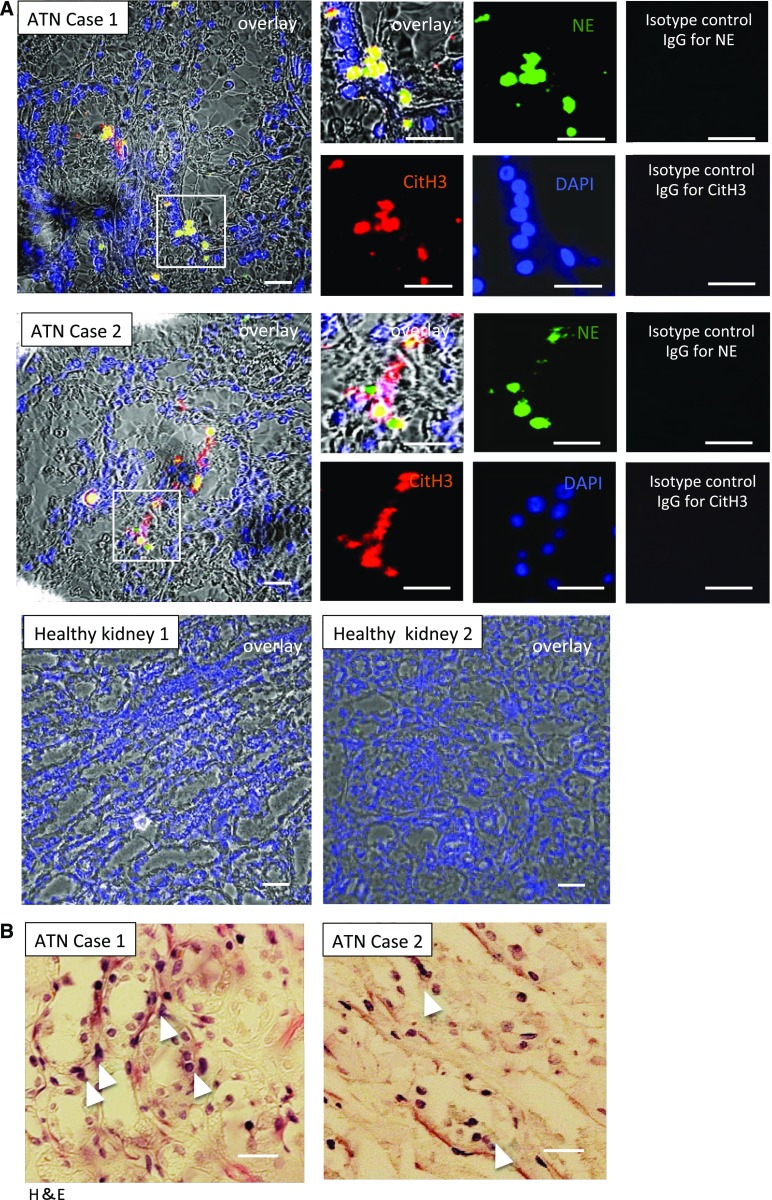
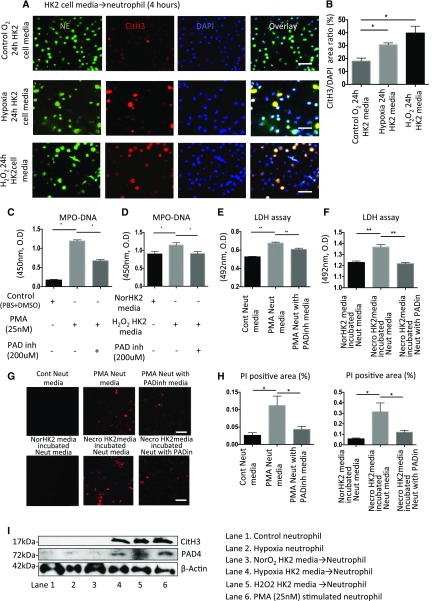
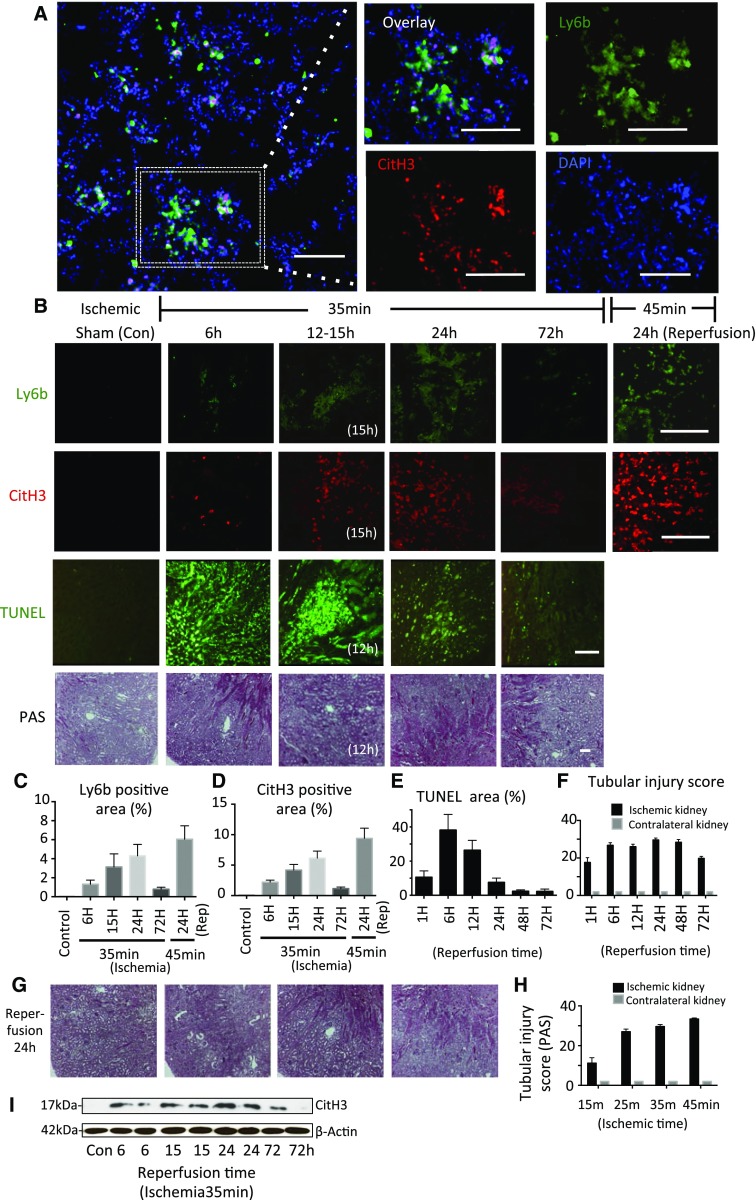
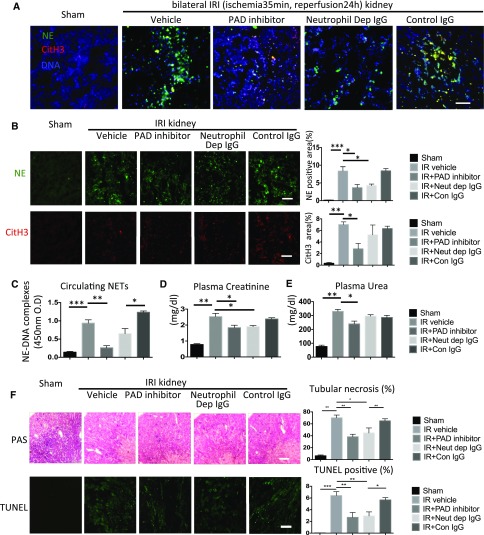
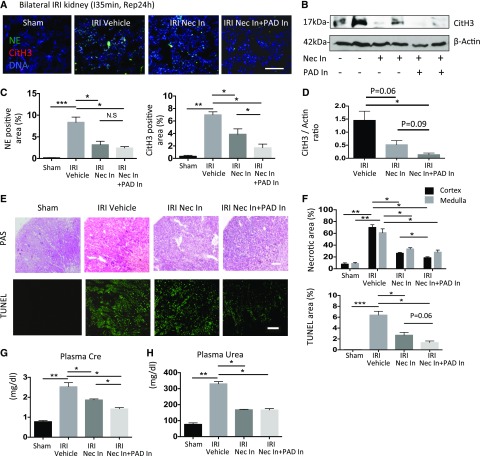
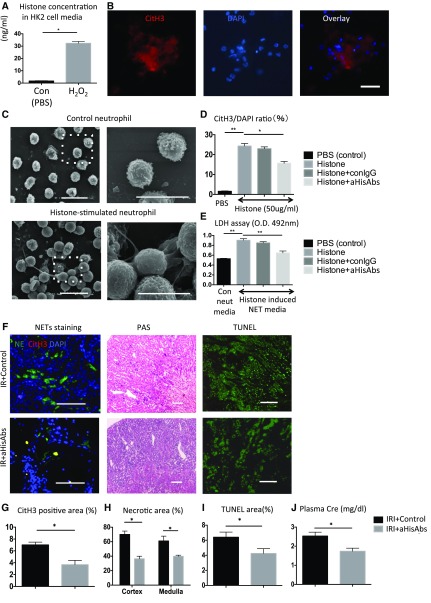
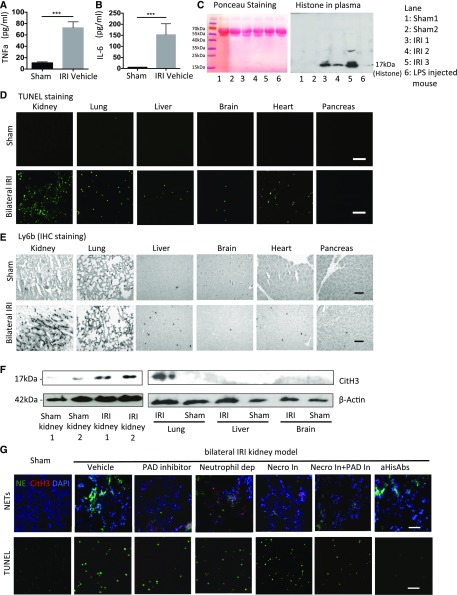
Severe AKI is often associated with multiorgan dysfunction, but the mechanisms of this remote tissue injury are unknown. We hypothesized that renal necroinflammation releases cytotoxic molecules that may cause remote organ damage. In hypoxia-induced tubular epithelial cell necrosis in vitro, histone secretion from ischemic tubular cells primed neutrophils to form neutrophil extracellular traps. These traps induced tubular epithelial cell death and stimulated neutrophil extracellular trap formation in fresh neutrophils. In vivo, ischemia-reperfusion injury in the mouse kidney induced tubular necrosis, which preceded the expansion of localized and circulating neutrophil extracellular traps and the increased expression of inflammatory and injury-related genes. Pretreatment with inhibitors of neutrophil extracellular trap formation reduced kidney injury. Dual inhibition of neutrophil trap formation and tubular cell necrosis had an additive protective effect. Moreover, pretreatment with antihistone IgG suppressed ischemia-induced neutrophil extracellular trap formation and renal injury. Renal ischemic injury also increased the levels of circulating histones, and we detected neutrophil infiltration and TUNEL-positive cells in the lungs, liver, brain, and heart along with neutrophil extracellular trap accumulation in the lungs. Inhibition of neutrophil extracellular trap formation or of circulating histones reduced these effects as well. These data suggest that tubular necrosis and neutrophil extracellular trap formation accelerate kidney damage and remote organ dysfunction through cytokine and histone release and identify novel molecular targets to limit renal necroinflammation and multiorgan failure.
Keywords: AKI related remote organ injury; Ischemic reperfusion injury; NETs; necroinflammation; tubular cell death.
Copyright © 2017 by the American Society of Nephrology.
Figures
Comment in
-
Neutrophil Extracellular Traps Orchestrate Necroinflammation.J Am Soc Nephrol. 2017 Jun;28(6):1670-1672. doi: 10.1681/ASN.2017010064. Epub 2017 Feb 23. J Am Soc Nephrol. 2017. PMID: 28232618 Free PMC article. No abstract available.
References
-
- Sharfuddin AA, Molitoris BA: Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol 7: 189–200, 2011 - PubMed
-
- Linkermann A, Stockwell BR, Krautwald S, Anders HJ: Regulated cell death and inflammation: An auto-amplification loop causes organ failure. Nat Rev Immunol 14: 759–767, 2014 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
