

DNA Polymerase Beta Germline Variant Confers Cellular Response to Cisplatin Therapy
- PMID: 28074003
- PMCID: PMC5334281
- DOI: 10.1158/1541-7786.MCR-16-0227-T
DNA Polymerase Beta Germline Variant Confers Cellular Response to Cisplatin Therapy
Abstract
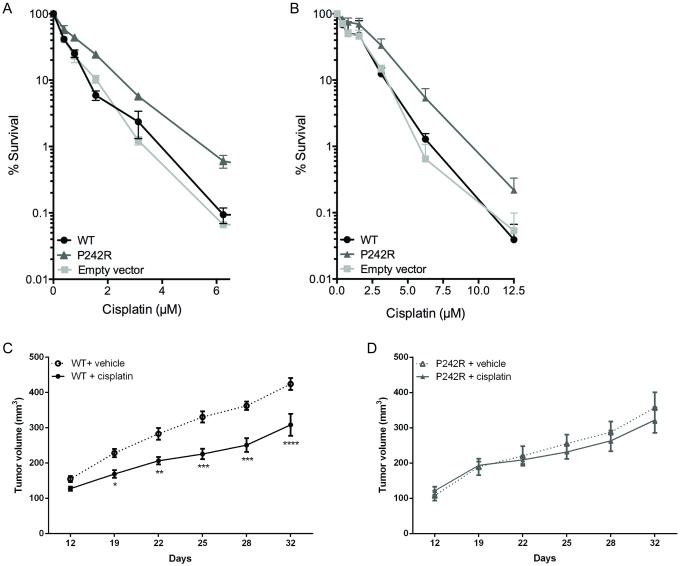
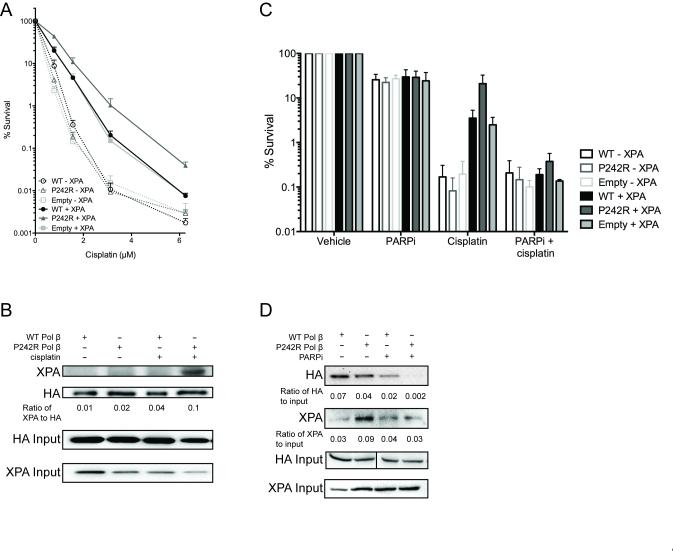
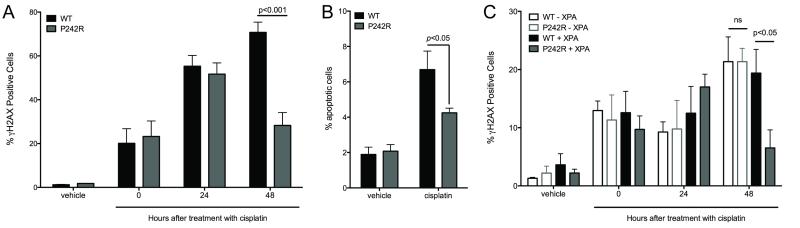
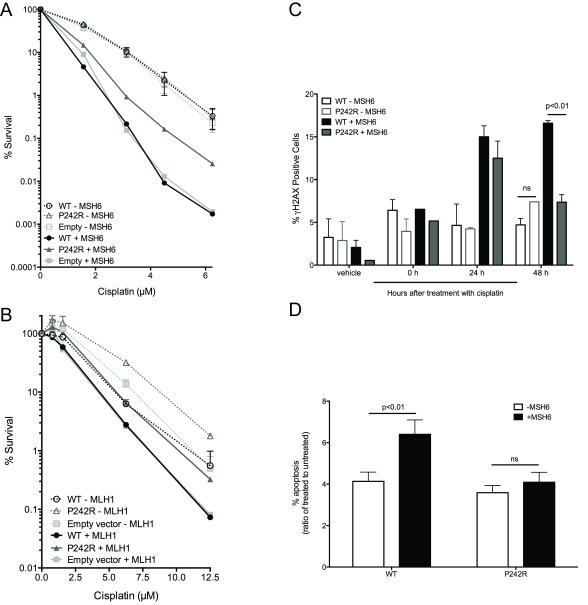
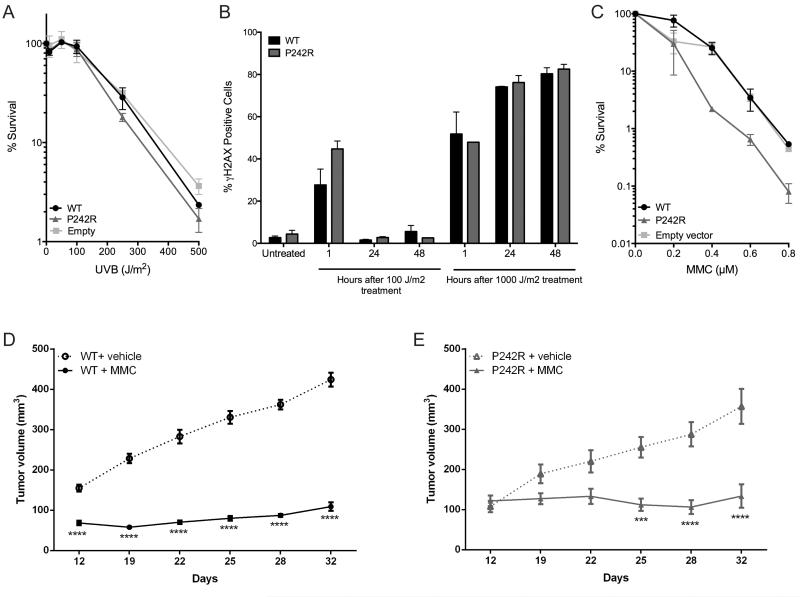
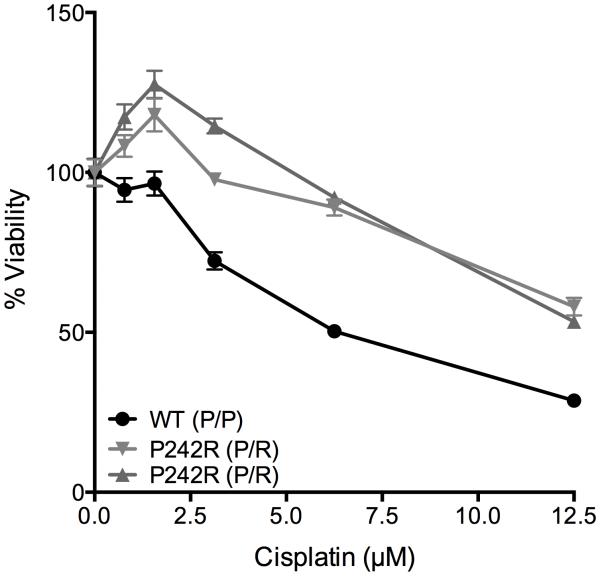
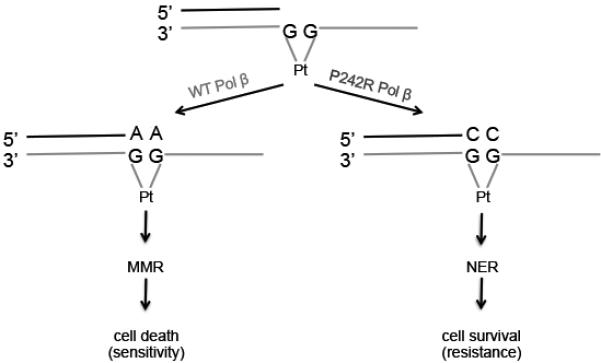
Resistance to cancer chemotherapies leads to deadly consequences, yet current research focuses only on the roles of somatically acquired mutations in this resistance. The mutational status of the germline is also likely to play a role in the way cells respond to chemotherapy. The carrier status for the POLB rs3136797 germline mutation encoding P242R DNA polymerase beta (Pol β) is associated with poor prognosis for lung cancer, specifically in response to treatment with cisplatin. Here, it is revealed that the P242R mutation is sufficient to promote resistance to cisplatin in human cells and in mouse xenografts. Mechanistically, P242R Pol β acts as a translesion polymerase and prefers to insert the correct nucleotide opposite cisplatin intrastrand cross-links, leading to the activation of the nucleotide excision repair (NER) pathway, removal of crosslinks, and resistance to cisplatin. In contrast, wild-type (WT) Pol β preferentially inserts the incorrect nucleotide initiating mismatch repair and cell death. Importantly, in a mouse xenograft model, tumors derived from lung cancer cells expressing WT Pol β displayed a slower rate of growth when treated with cisplatin, whereas tumors expressing P242R Pol β had no response to cisplatin. Pol β is critical for mediating crosstalk in response to cisplatin. The current data strongly suggest that the status of Pol β influences cellular responses to crosslinking agents and that Pol β is a promising biomarker to predict responses to specific chemotherapies. Finally, these results highlight that the genetic status of the germline is a critical factor in the response to cancer treatment.Implications: Pol β has prognostic biomarker potential in the treatment of cancer with cisplatin and perhaps other intrastrand crosslinking agents. Mol Cancer Res; 15(3); 269-80. ©2017 AACR.
©2017 American Association for Cancer Research.
Figures
References
-
- Weiss RB, Christian MC. New cisplatin analogues in development. A review. Drugs. 1993;46(3):360–77. - PubMed
-
- Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4(4):307–20. - PubMed
-
- Eastman A. The formation, isolation and characterization of DNA adducts produced by anticancer platinum complexes. Pharmacol Ther. 1987;34(2):155–66. - PubMed
-
- Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutat Res. 2001;478(1-2):23–43. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
