

Glioma: experimental models and reality
- PMID: 28074274
- PMCID: PMC5250671
- DOI: 10.1007/s00401-017-1671-4
Glioma: experimental models and reality
Abstract
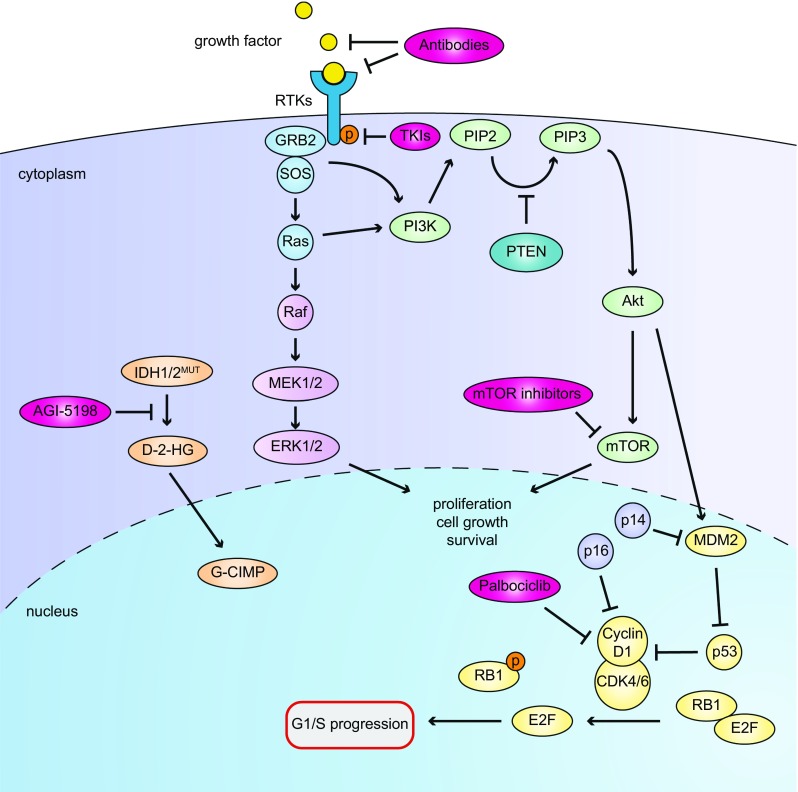
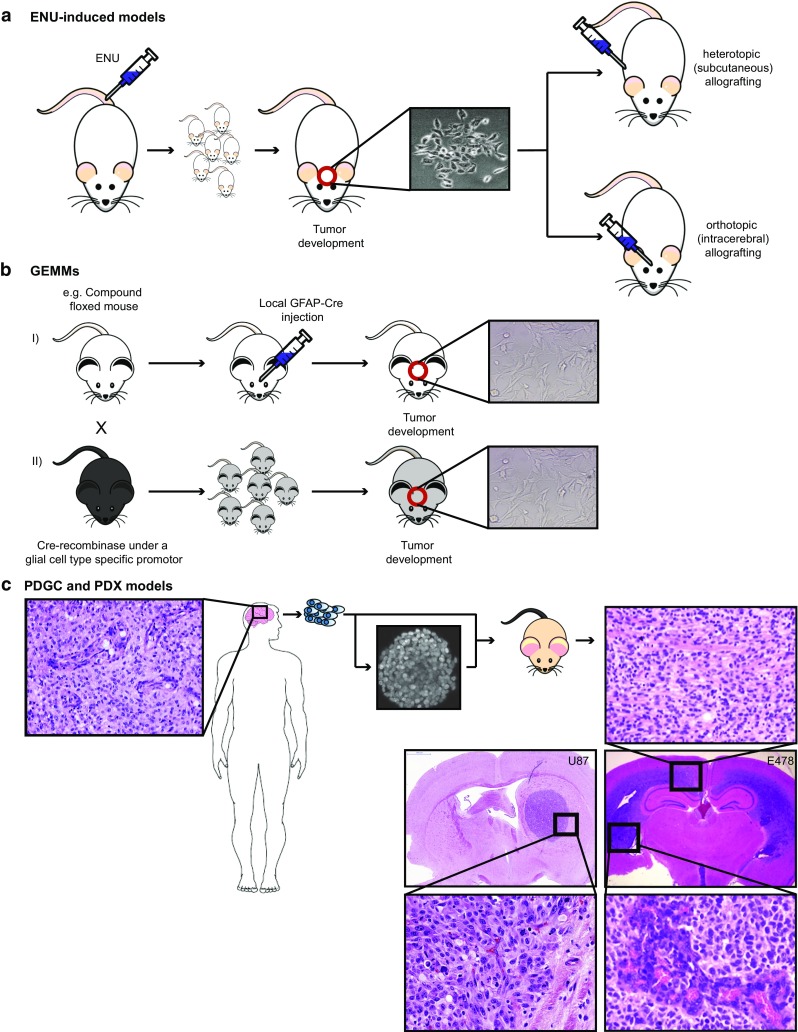
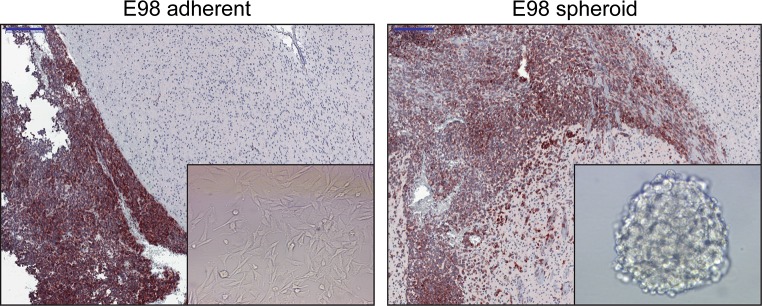
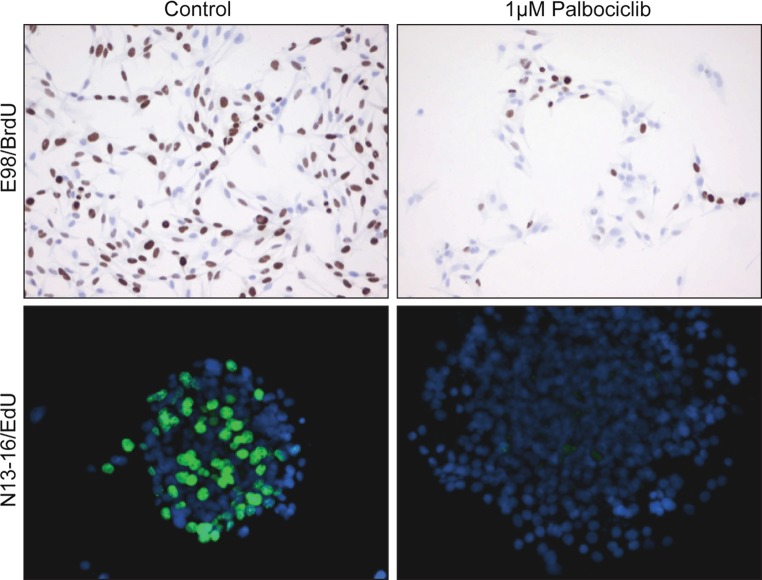
In theory, in vitro and in vivo models for human gliomas have great potential to not only enhance our understanding of glioma biology, but also to facilitate the development of novel treatment strategies for these tumors. For reliable prediction and validation of the effects of different therapeutic modalities, however, glioma models need to comply with specific and more strict demands than other models of cancer, and these demands are directly related to the combination of genetic aberrations and the specific brain micro-environment gliomas grow in. This review starts with a brief introduction on the pathological and molecular characteristics of gliomas, followed by an overview of the models that have been used in the last decades in glioma research. Next, we will discuss how these models may play a role in better understanding glioma development and especially in how they can aid in the design and optimization of novel therapies. The strengths and weaknesses of the different models will be discussed in light of genotypic, phenotypic and metabolic characteristics of human gliomas. The last part of this review provides some examples of how therapy experiments using glioma models can lead to deceptive results when such characteristics are not properly taken into account.
Conflict of interest statement
Compliance with ethical standards Financial support KL is funded by KWF Grant UvA2014-6839. This work was in part supported by a Grant from Stichting StopHersentumoren and by Radboudumc. RGWV is supported by Grants from the National Brain Tumor Society and NIH/NCI R01 CA190121.
Figures
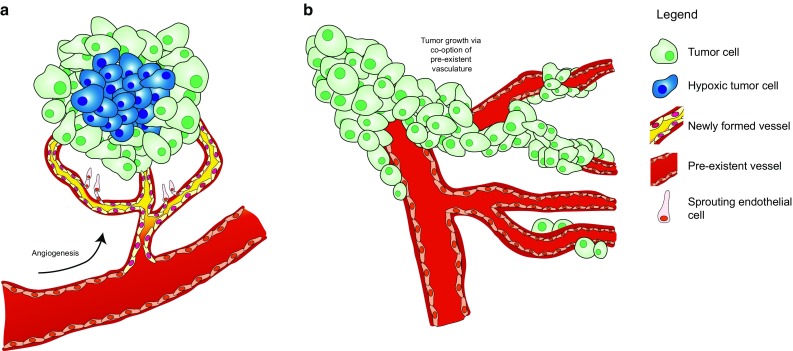
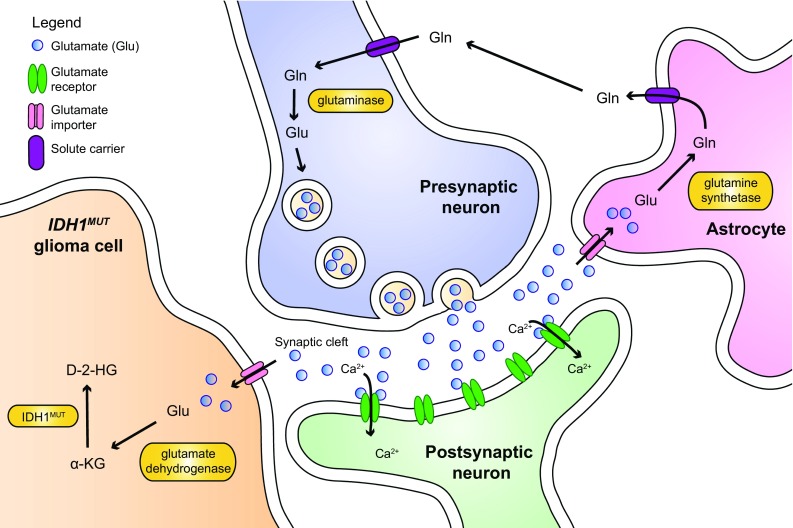
References
-
- Allen M, Bjerke M, Edlund H, Nelander S, Westermark B (2016) Origin of the U87MG glioma cell line: Good news and bad news. Sci Transl Med 8:354re353. doi:10.1126/scitranslmed.aaf6853 - PubMed
-
- Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016 - PubMed
-
- Ausman JI, Shapiro WR, Rall DP. Studies on the chemotherapy of experimental brain tumors: development of an experimental model. Cancer Res. 1970;30:2394–2400. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
