

MLL-AF4 Spreading Identifies Binding Sites that Are Distinct from Super-Enhancers and that Govern Sensitivity to DOT1L Inhibition in Leukemia
- PMID: 28076791
- PMCID: PMC5263239
- DOI: 10.1016/j.celrep.2016.12.054
MLL-AF4 Spreading Identifies Binding Sites that Are Distinct from Super-Enhancers and that Govern Sensitivity to DOT1L Inhibition in Leukemia
Abstract
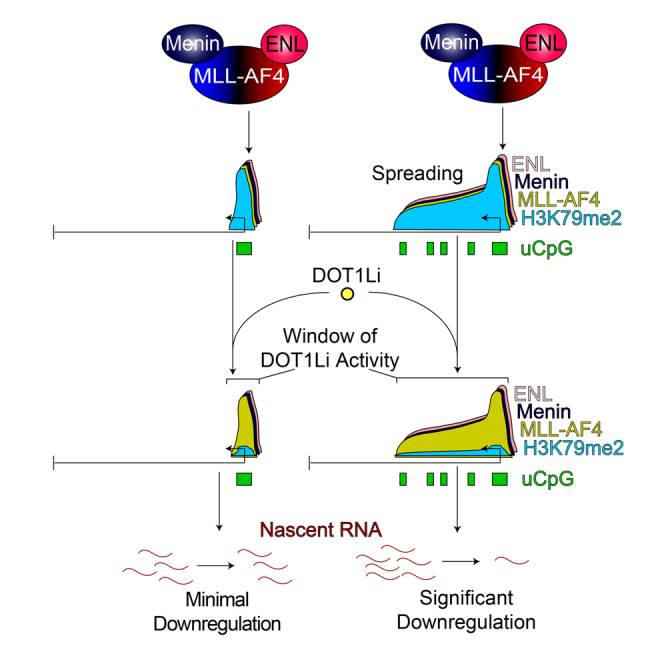
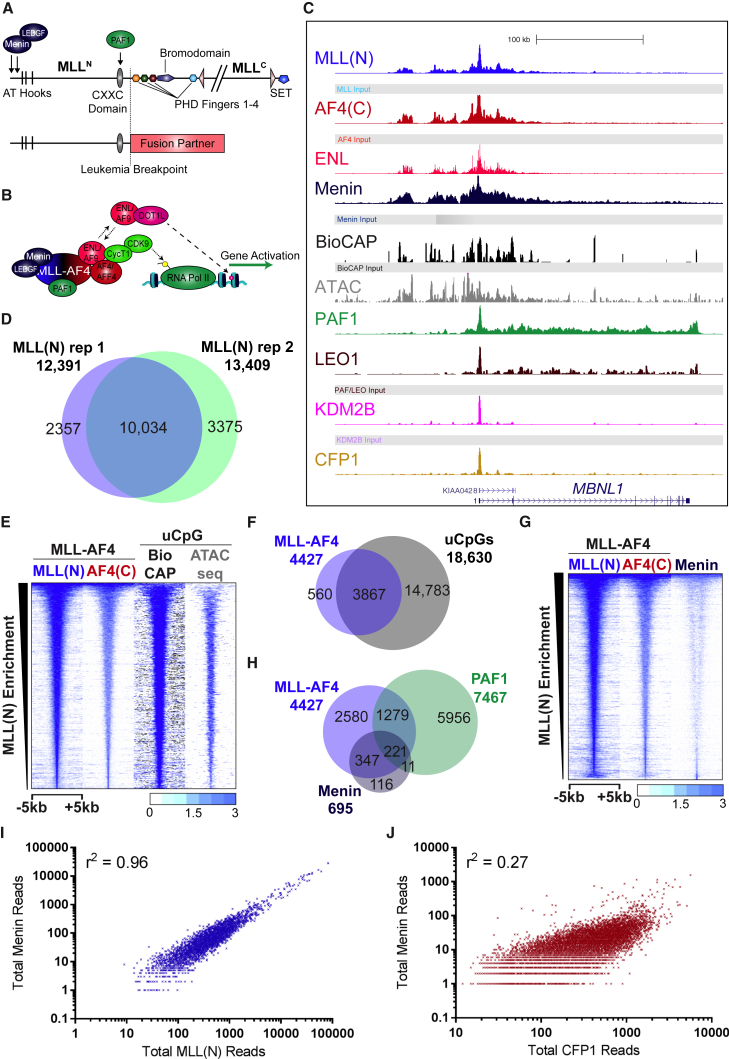
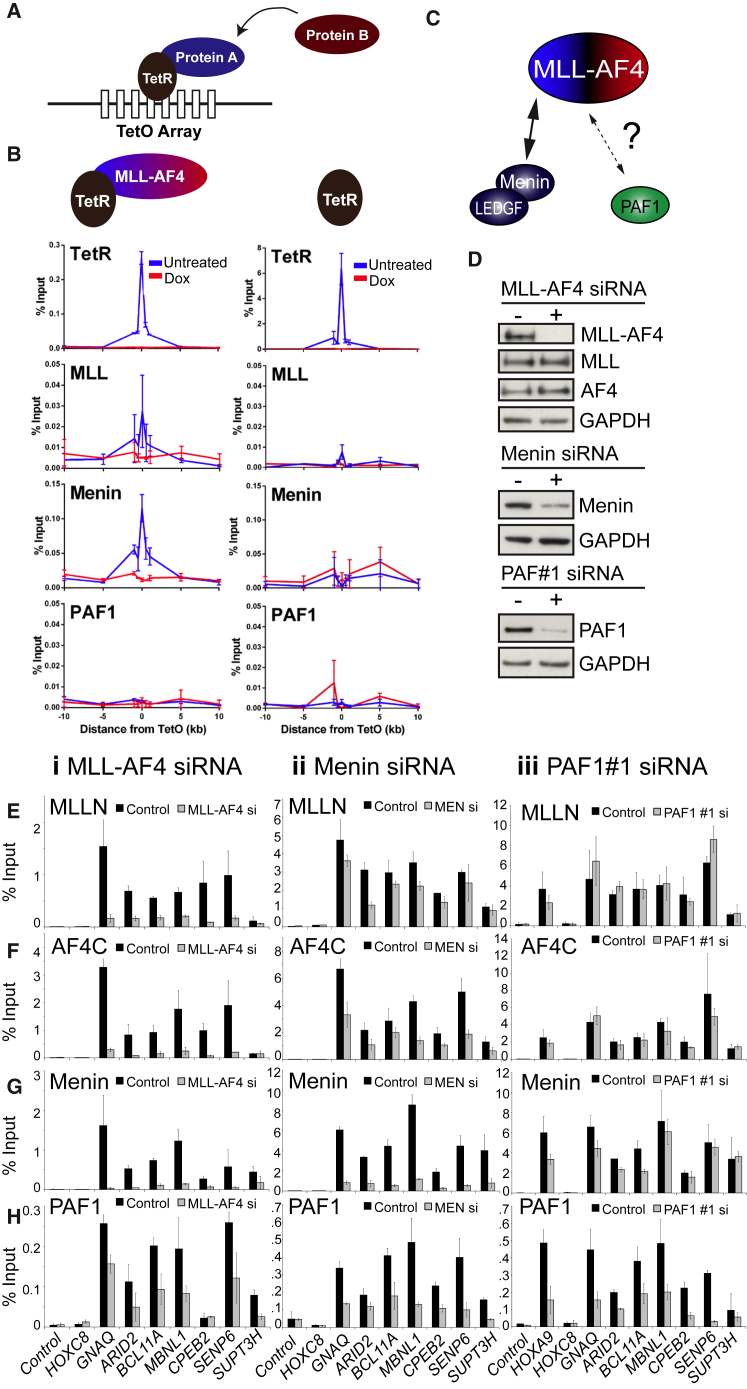
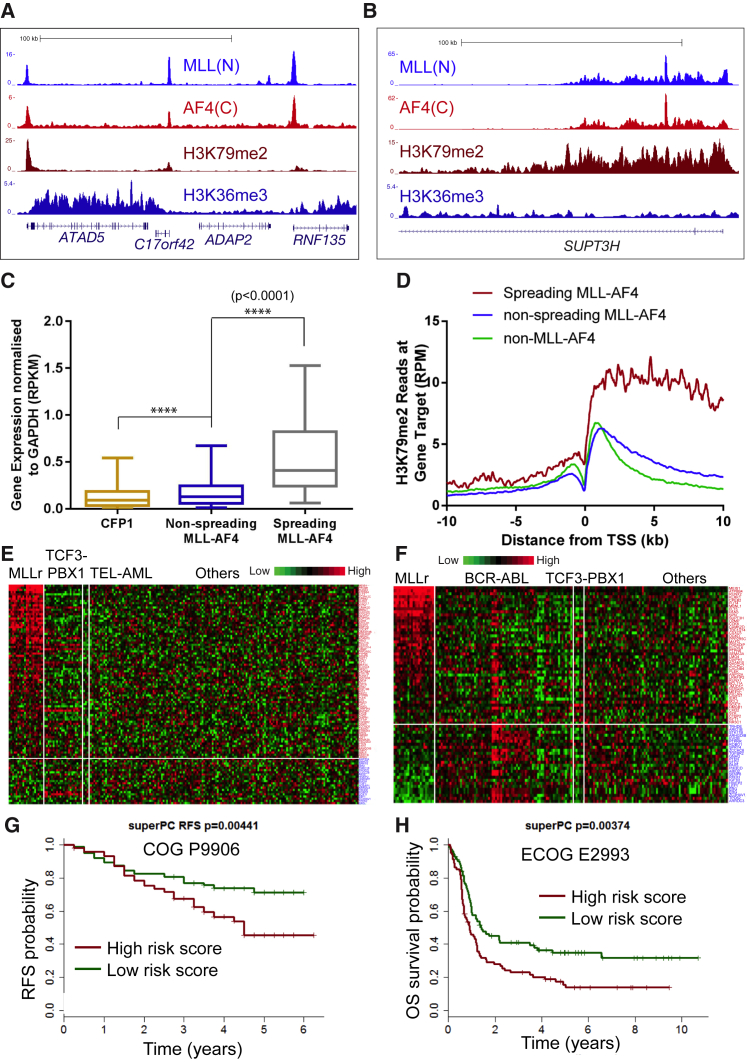
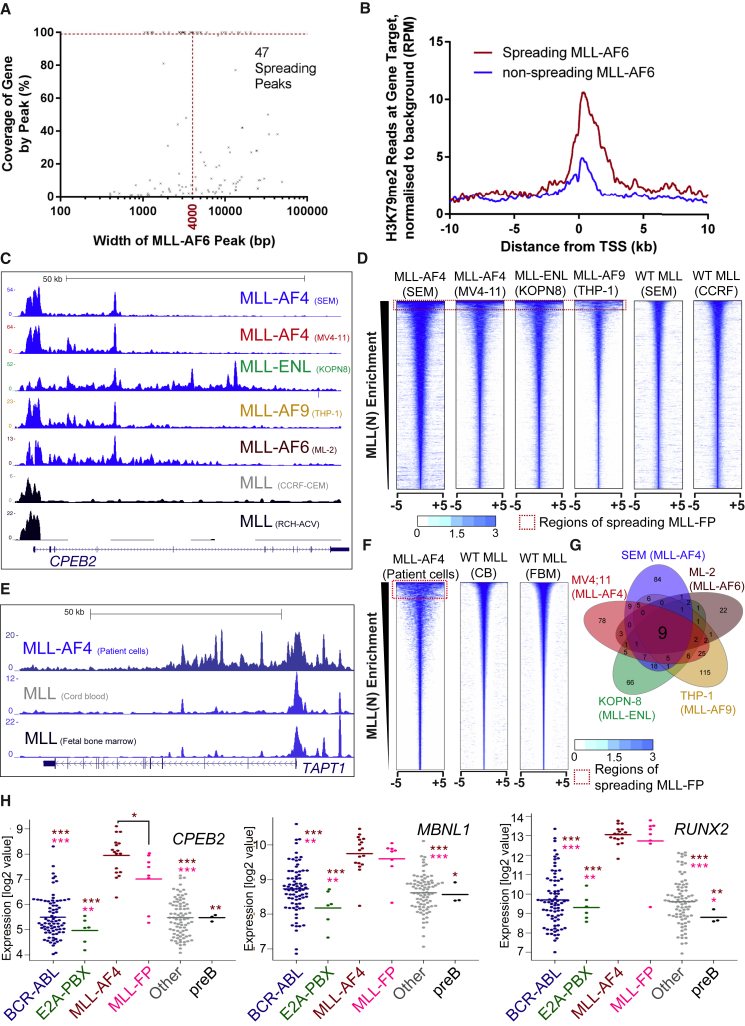
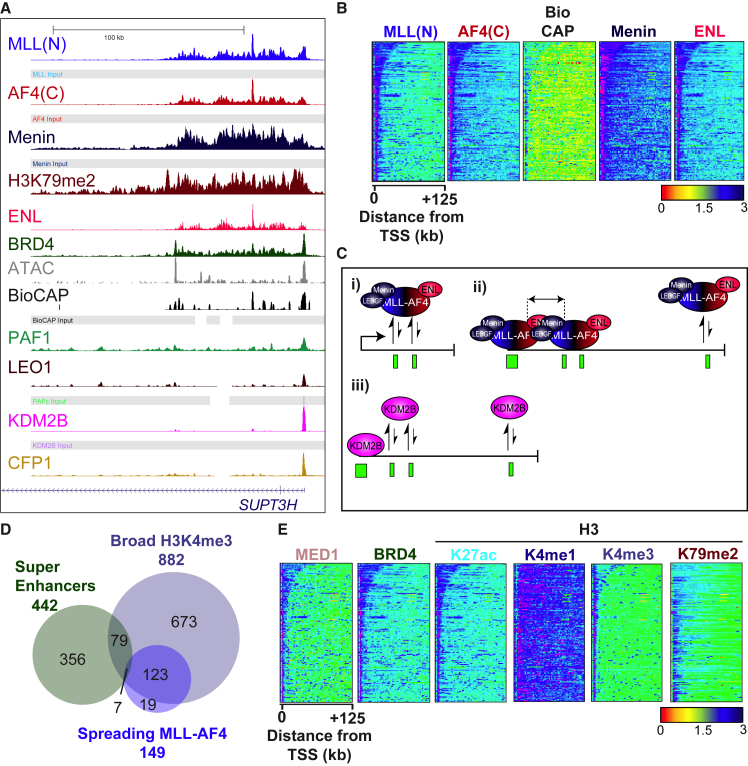
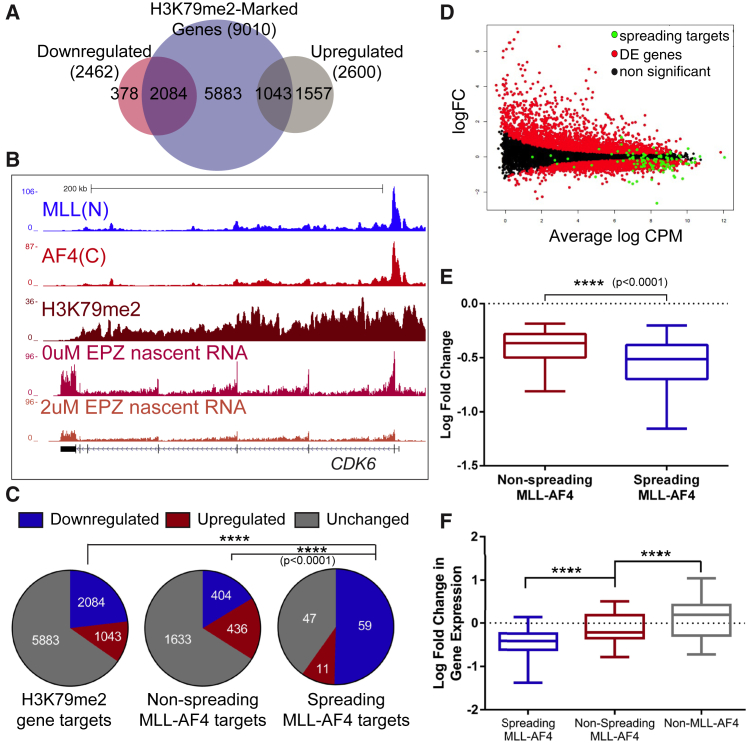
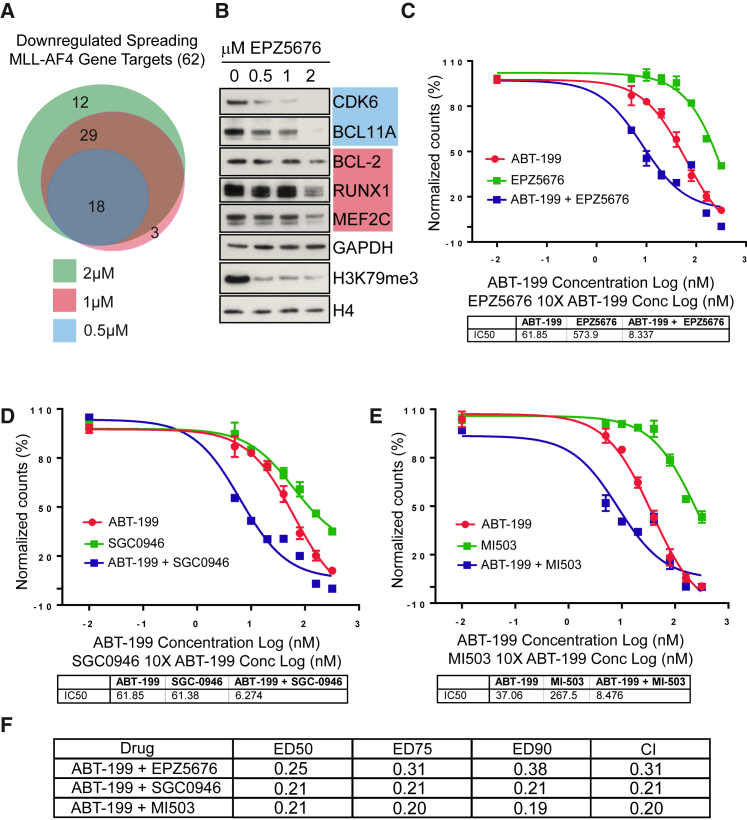
Understanding the underlying molecular mechanisms of defined cancers is crucial for effective personalized therapies. Translocations of the mixed-lineage leukemia (MLL) gene produce fusion proteins such as MLL-AF4 that disrupt epigenetic pathways and cause poor-prognosis leukemias. Here, we find that at a subset of gene targets, MLL-AF4 binding spreads into the gene body and is associated with the spreading of Menin binding, increased transcription, increased H3K79 methylation (H3K79me2/3), a disruption of normal H3K36me3 patterns, and unmethylated CpG regions in the gene body. Compared to other H3K79me2/3 marked genes, MLL-AF4 spreading gene expression is downregulated by inhibitors of the H3K79 methyltransferase DOT1L. This sensitivity mediates synergistic interactions with additional targeted drug treatments. Therefore, epigenetic spreading and enhanced susceptibility to epidrugs provides a potential marker for better understanding combination therapies in humans.
Keywords: DOT1L; H3K79me2; MLL; MLL-AF4; drug combination therapy; epigenetic spreading; epigenetic therapy; leukemia.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Benito J.M., Godfrey L., Kojima K., Hogdal L., Wunderlich M., Geng H., Marzo I., Harutyunyan K.G., Golfman L., North P. MLL-rearranged acute lymphoblastic leukemias activate BCL-2 through H3K79 methylation and are sensitive to the BCL-2-specific antagonist ABT-199. Cell Rep. 2015;13:2715–2727. - PMC - PubMed
MeSH terms
Substances
Grants and funding
- MC_UU_00016/6/MRC_/Medical Research Council/United Kingdom
- 099175/Z/12/Z/WT_/Wellcome Trust/United Kingdom
- MC_PC_15004/MRC_/Medical Research Council/United Kingdom
- 098024/Z/11/Z/WT_/Wellcome Trust/United Kingdom
- MC_UU_00016/13/MRC_/Medical Research Council/United Kingdom
- G0700089/MRC_/Medical Research Council/United Kingdom
- 090532/Z/09/Z/WT_/Wellcome Trust/United Kingdom
- 12788/CRUK_/Cancer Research UK/United Kingdom
- MC_UU_12009/14/MRC_/Medical Research Council/United Kingdom
- C27943/A12788/CRUK_/Cancer Research UK/United Kingdom
- MC_UU_12009/6/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
