

High-throughput sequencing of the T-cell receptor repertoire: pitfalls and opportunities
- PMID: 28077404
- PMCID: PMC6054146
- DOI: 10.1093/bib/bbw138
High-throughput sequencing of the T-cell receptor repertoire: pitfalls and opportunities
Abstract
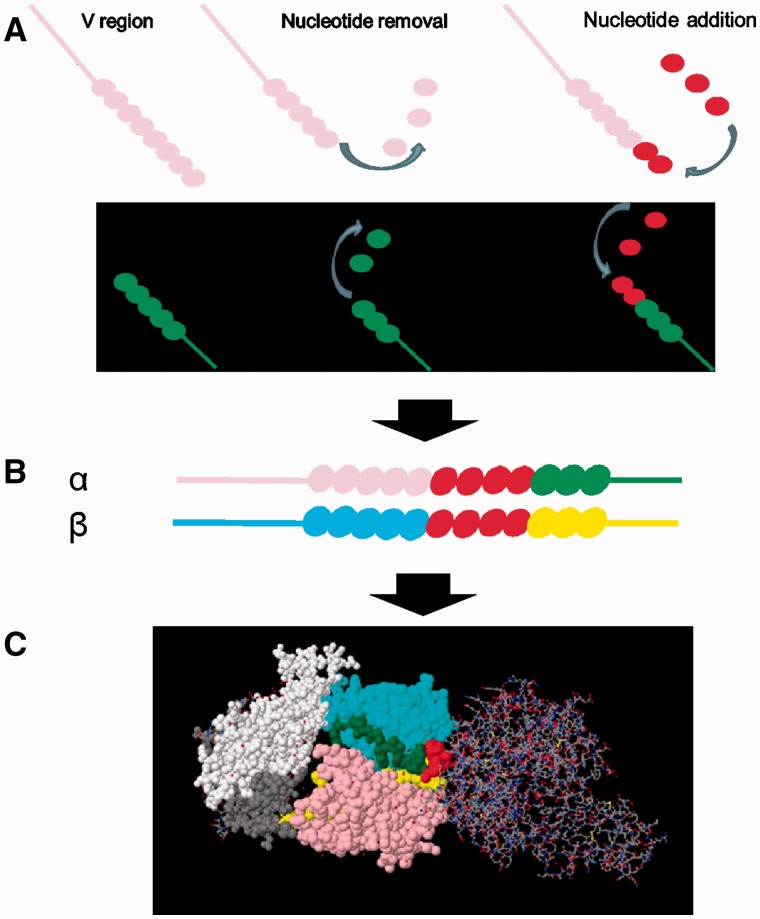
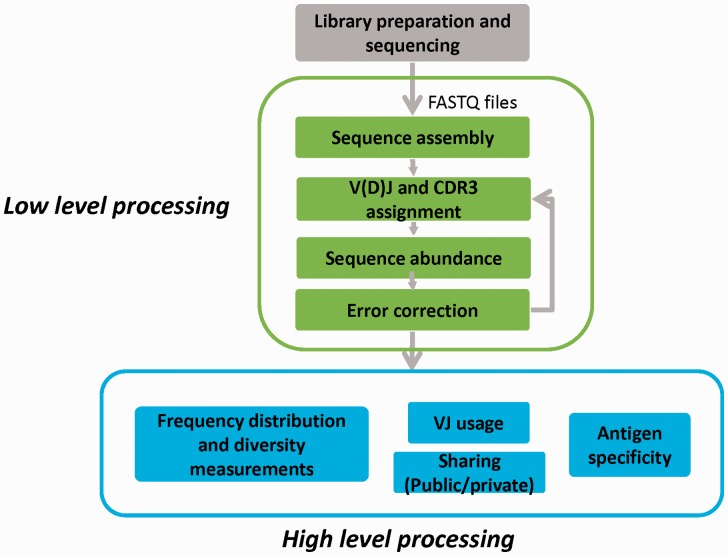
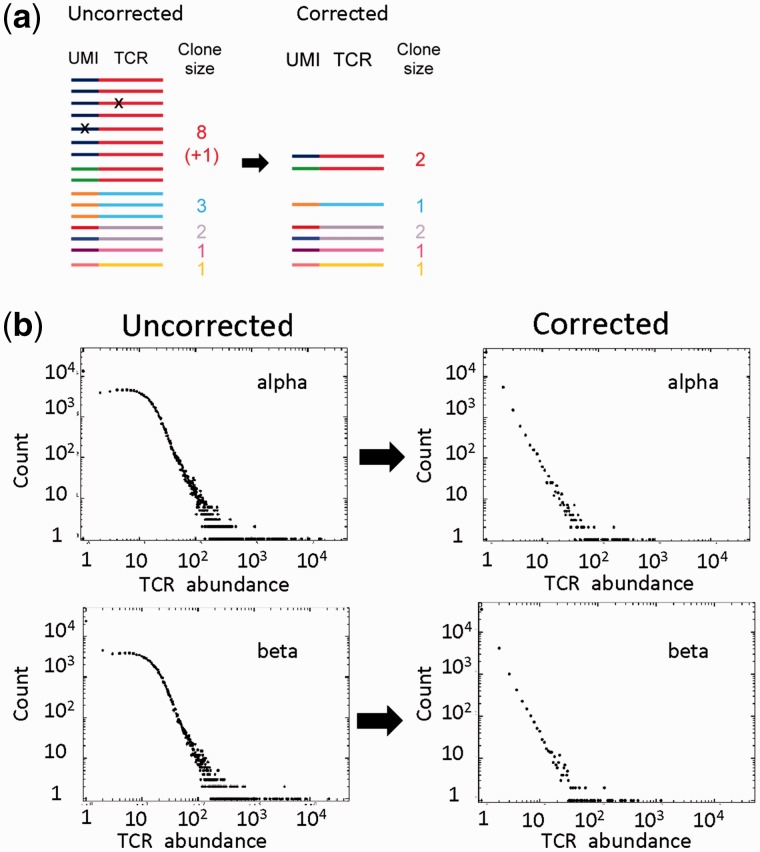
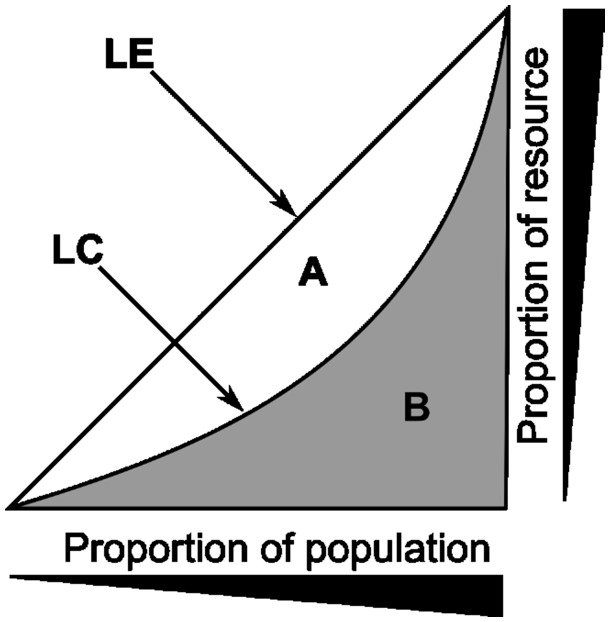
T-cell specificity is determined by the T-cell receptor, a heterodimeric protein coded for by an extremely diverse set of genes produced by imprecise somatic gene recombination. Massively parallel high-throughput sequencing allows millions of different T-cell receptor genes to be characterized from a single sample of blood or tissue. However, the extraordinary heterogeneity of the immune repertoire poses significant challenges for subsequent analysis of the data. We outline the major steps in processing of repertoire data, considering low-level processing of raw sequence files and high-level algorithms, which seek to extract biological or pathological information. The latest generation of bioinformatics tools allows millions of DNA sequences to be accurately and rapidly assigned to their respective variable V and J gene segments, and to reconstruct an almost error-free representation of the non-templated additions and deletions that occur. High-level processing can measure the diversity of the repertoire in different samples, quantify V and J usage and identify private and public T-cell receptors. Finally, we discuss the major challenge of linking T-cell receptor sequence to function, and specifically to antigen recognition. Sophisticated machine learning algorithms are being developed that can combine the paradoxical degeneracy and cross-reactivity of individual T-cell receptors with the specificity of the overall T-cell immune response. Computational analysis will provide the key to unlock the potential of the T-cell receptor repertoire to give insight into the fundamental biology of the adaptive immune system and to provide powerful biomarkers of disease.
Figures
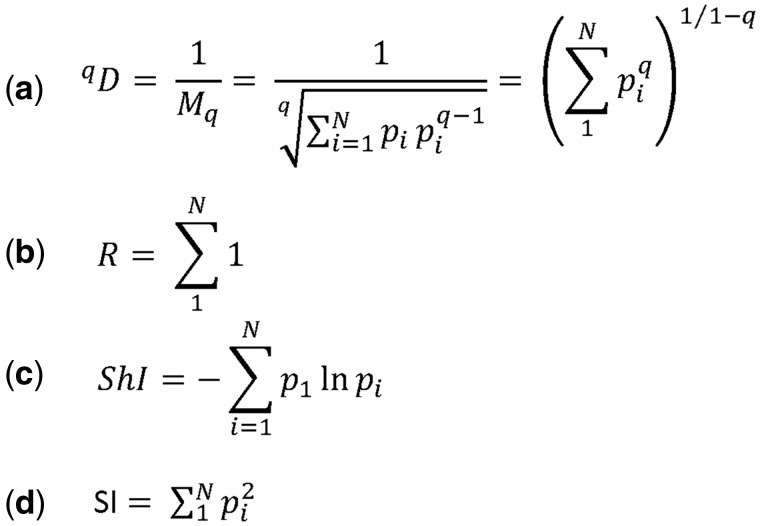
References
-
- Alt FW, Oltz EM, Young F, et al.VDJ recombination. Immunol Today 1992; 13:306–14. - PubMed
-
- Bernstein HJ. Recent changes to RasMol, recombining the variants. Trends Biochem Sci 2000; 25:453–5. - PubMed
-
- Vanhanen R, Heikkilä N, Aggarwal K, et al.T cell receptor diversity in the human thymus. Mol Immunol 2016; 76:116–22. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
