

Prion disease: experimental models and reality
- PMID: 28084518
- PMCID: PMC5250673
- DOI: 10.1007/s00401-017-1670-5
Prion disease: experimental models and reality
Abstract
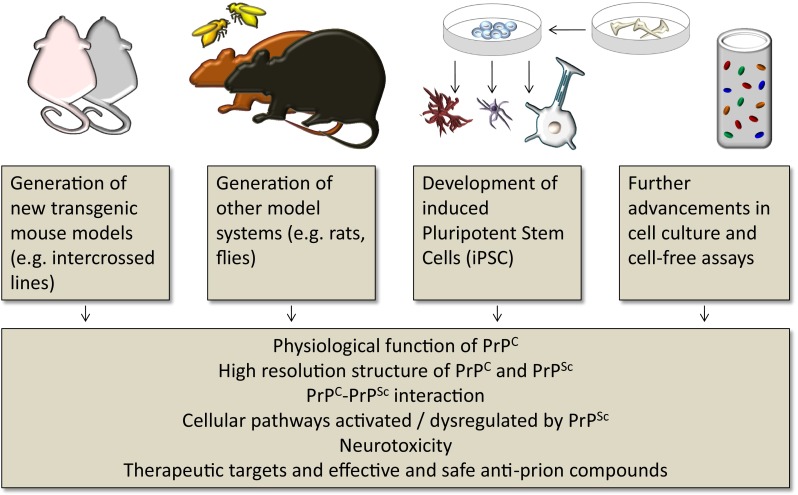
The understanding of the pathogenesis and mechanisms of diseases requires a multidisciplinary approach, involving clinical observation, correlation to pathological processes, and modelling of disease mechanisms. It is an inherent challenge, and arguably impossible to generate model systems that can faithfully recapitulate all aspects of human disease. It is, therefore, important to be aware of the potentials and also the limitations of specific model systems. Model systems are usually designed to recapitulate only specific aspects of the disease, such as a pathological phenotype, a pathomechanism, or to test a hypothesis. Here, we evaluate and discuss model systems that were generated to understand clinical, pathological, genetic, biochemical, and epidemiological aspects of prion diseases. Whilst clinical research and studies on human tissue are an essential component of prion research, much of the understanding of the mechanisms governing transmission, replication, and toxicity comes from in vitro and in vivo studies. As with other neurodegenerative diseases caused by protein misfolding, the pathogenesis of prion disease is complex, full of conundra and contradictions. We will give here a historical overview of the use of models of prion disease, how they have evolved alongside the scientific questions, and how advancements in technologies have pushed the boundaries of our understanding of prion biology.
Figures
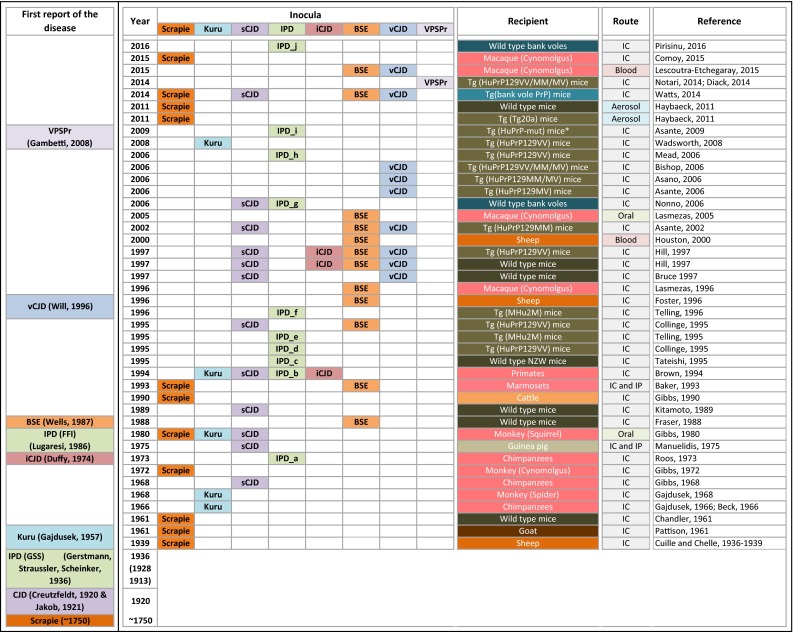

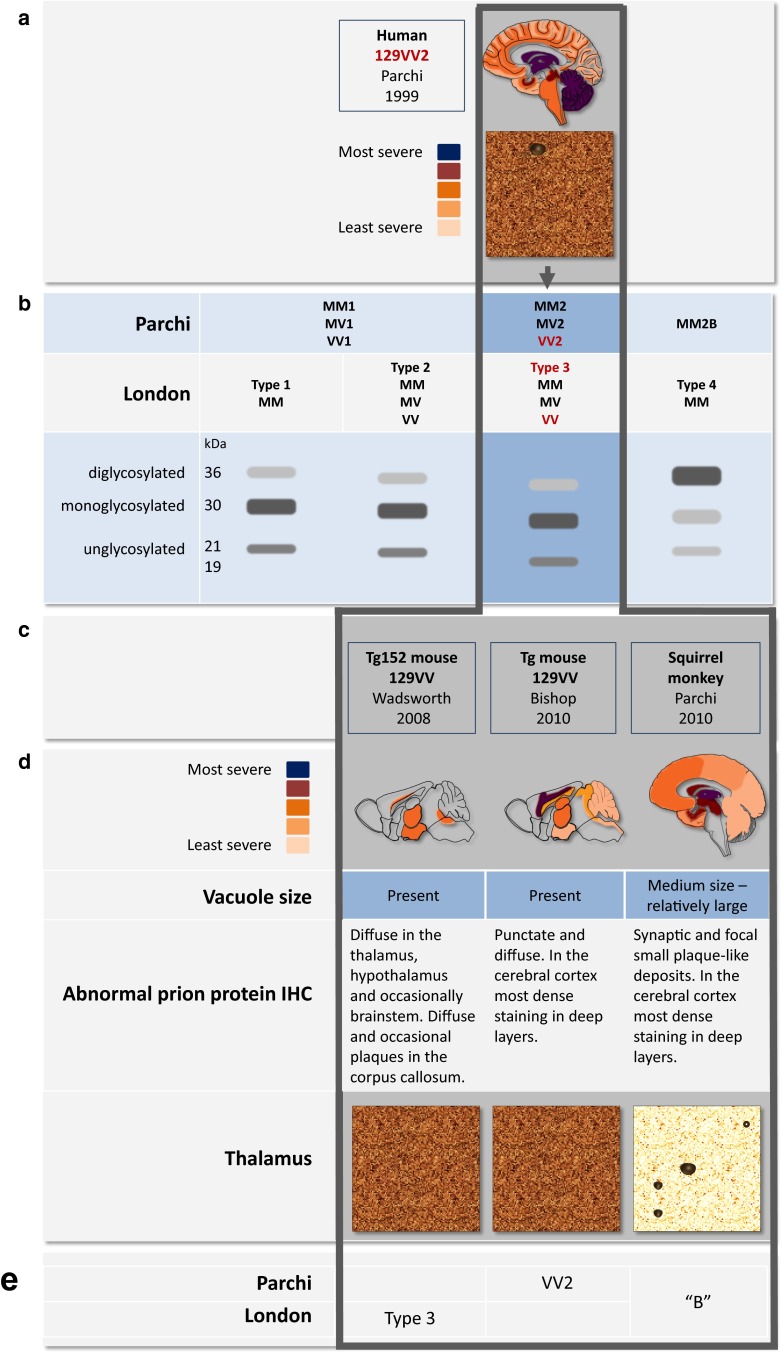
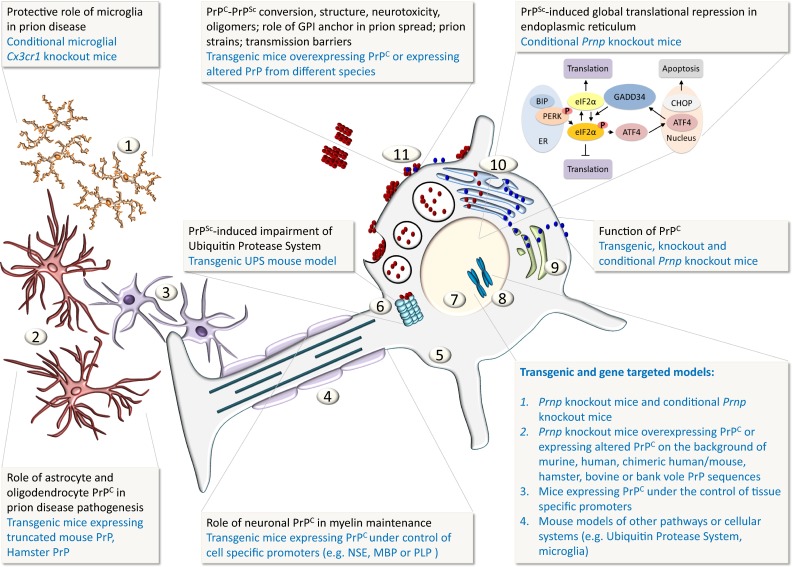
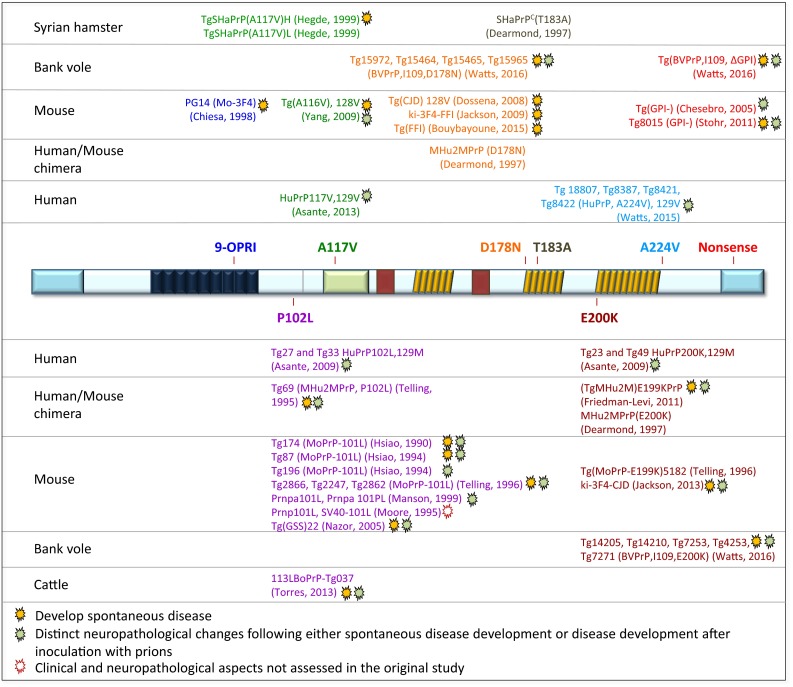
References
-
- Asante EA, Gowland I, Grimshaw A, Linehan JM, Smidak M, Houghton R, Osiguwa O, Tomlinson A, Joiner S, Brandner S, et al. Absence of spontaneous disease and comparative prion susceptibility of transgenic mice expressing mutant human prion proteins. J Gen Virol. 2009;90:546–558. doi: 10.1099/vir.0.007930-0. - DOI - PMC - PubMed
-
- Asante EA, Grimshaw A, Smidak M, Jakubcova T, Tomlinson A, Jeelani A, Hamdan S, Powell C, Joiner S, Linehan JM, et al. Transmission properties of human PrP 102L prions challenge the relevance of mouse models of GSS. PLoS Pathog. 2015;11:e1004953. doi: 10.1371/journal.ppat.1004953. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
