

Deletion of PRKAA triggers mitochondrial fission by inhibiting the autophagy-dependent degradation of DNM1L
- PMID: 28085543
- PMCID: PMC5324848
- DOI: 10.1080/15548627.2016.1263776
Deletion of PRKAA triggers mitochondrial fission by inhibiting the autophagy-dependent degradation of DNM1L
Abstract
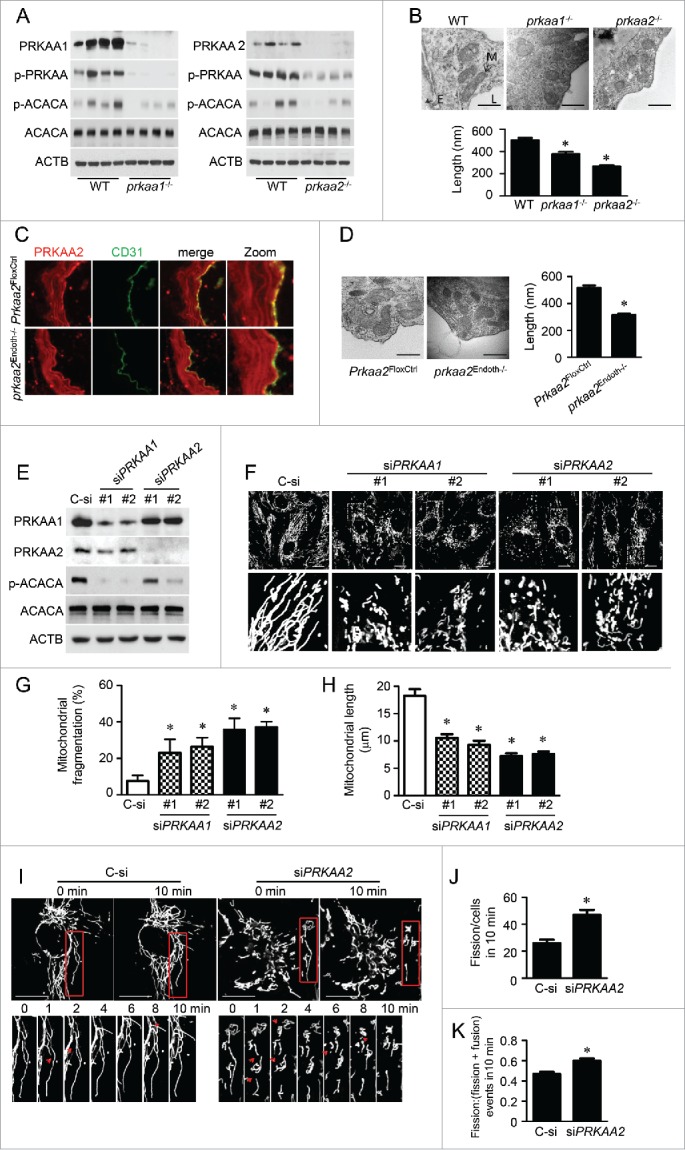
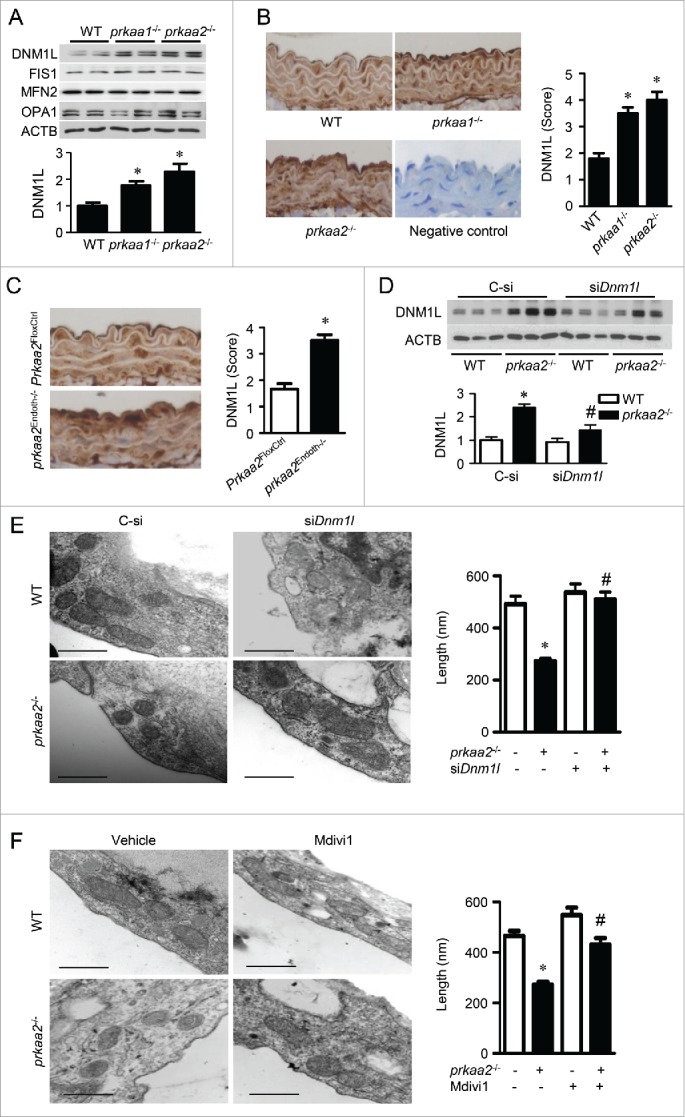
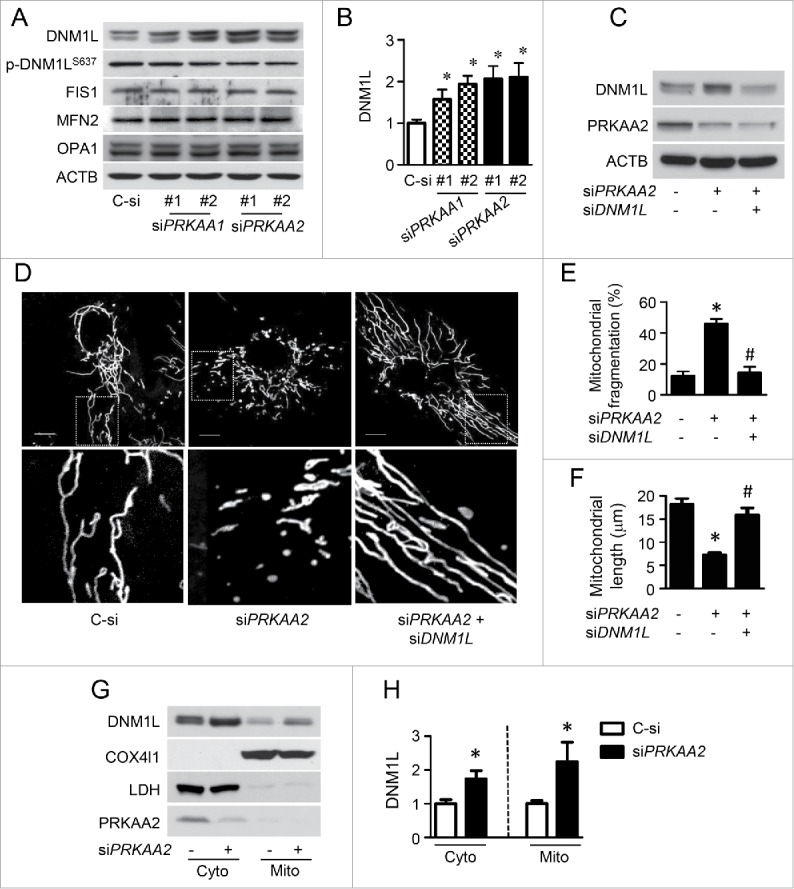
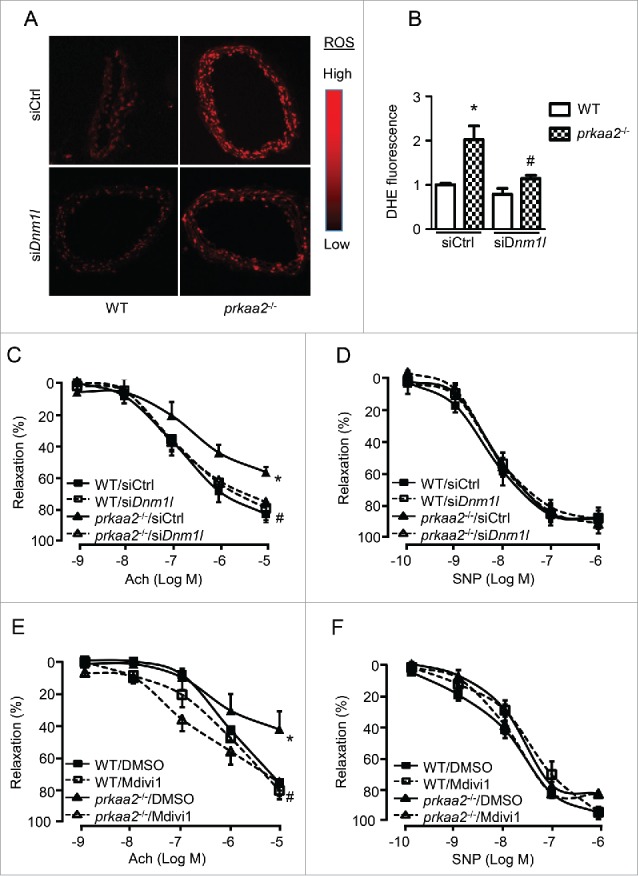
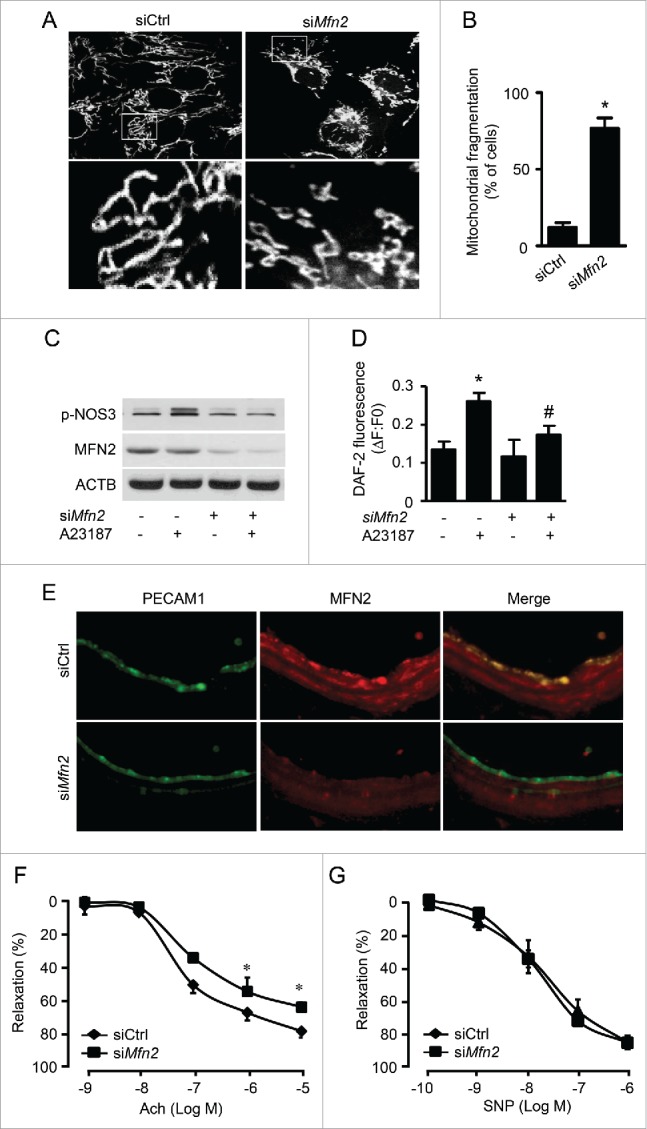
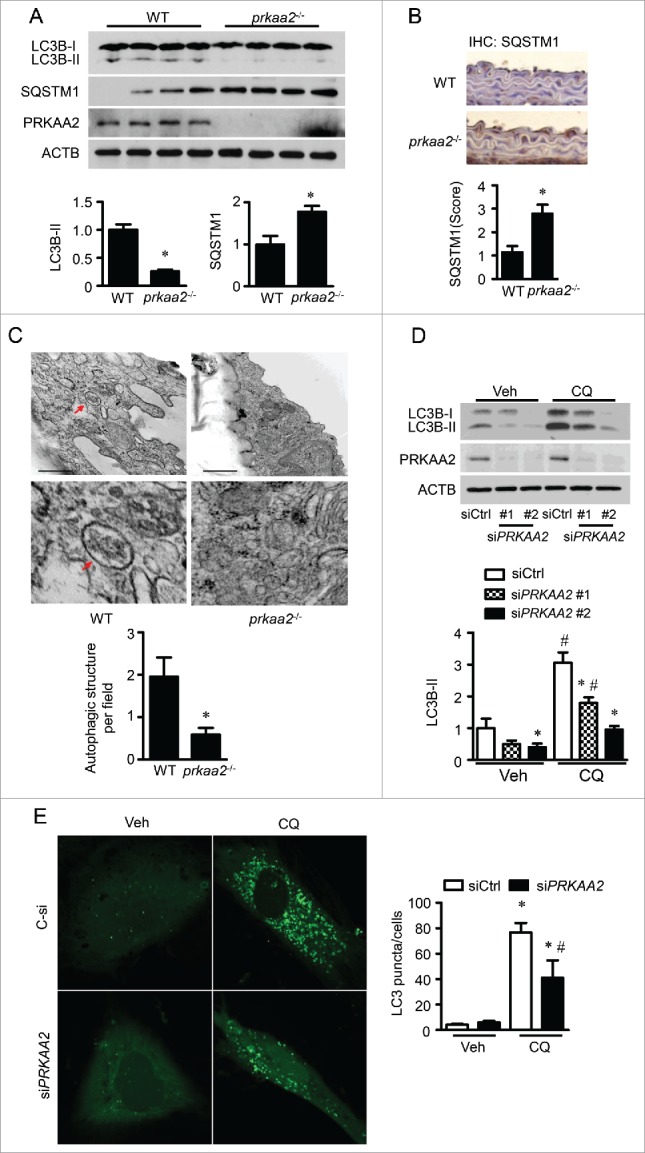
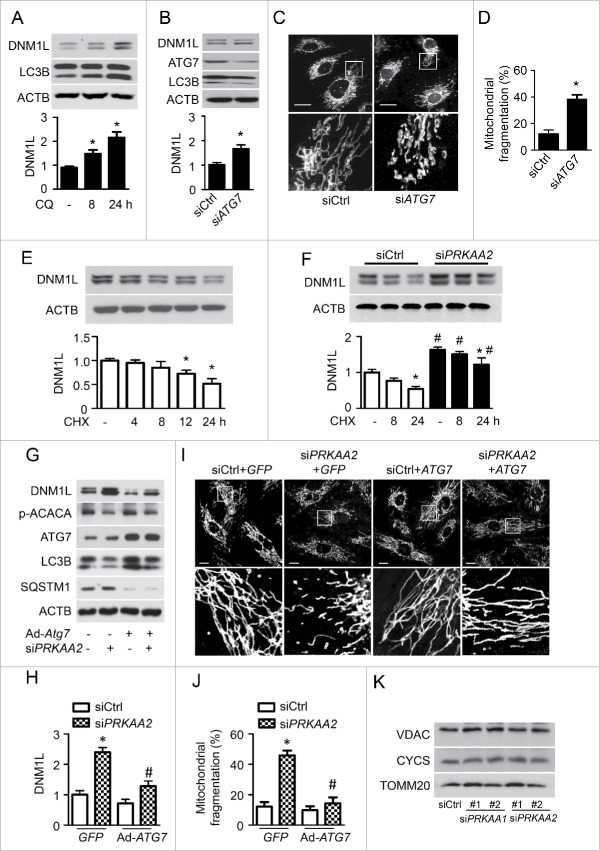
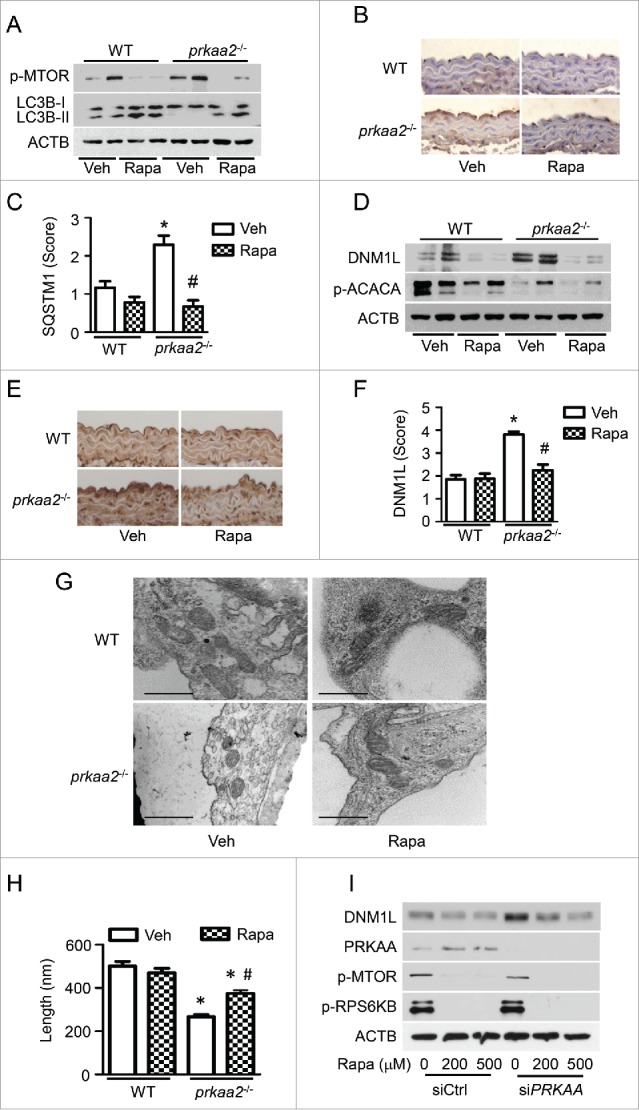
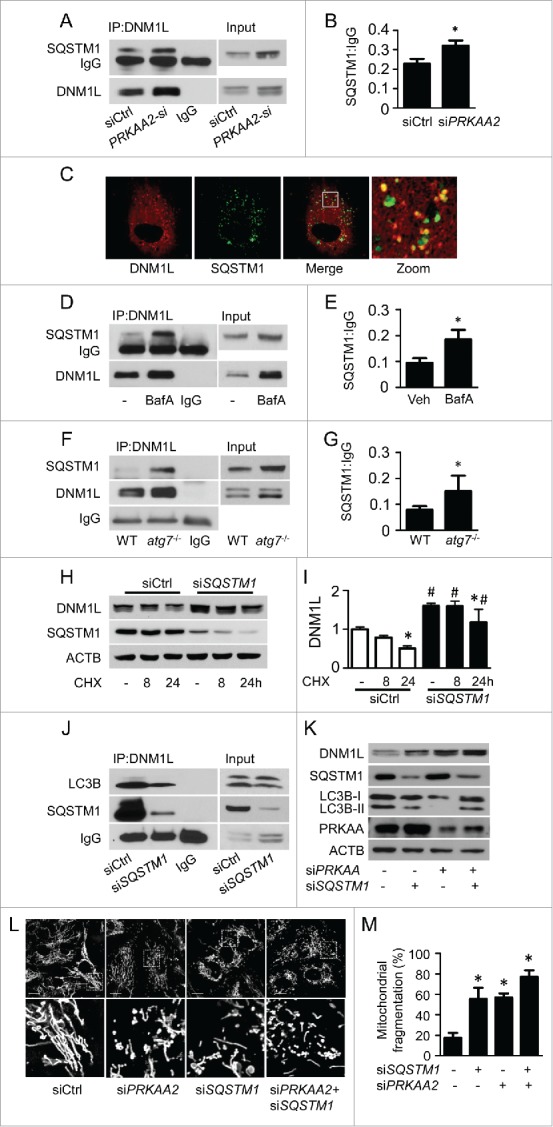
PRKAA (protein kinase, AMP-activated, α catalytic subunit) regulates mitochondrial biogenesis, function, and turnover. However, the molecular mechanisms by which PRKAA regulates mitochondrial dynamics remain poorly characterized. Here, we report that PRKAA regulated mitochondrial fission via the autophagy-dependent degradation of DNM1L (dynamin 1-like). Deletion of Prkaa1/AMPKα1 or Prkaa2/AMPKα2 resulted in defective autophagy, DNM1L accumulation, and aberrant mitochondrial fragmentation in the mouse aortic endothelium. Furthermore, autophagy inhibition by chloroquine treatment or ATG7 small interfering RNA (siRNA) transfection, upregulated DNM1L expression and triggered DNM1L-mediated mitochondrial fragmentation. In contrast, autophagy activation by overexpression of ATG7 or chronic administration of rapamycin, the MTOR inhibitor, promoted DNM1L degradation and attenuated mitochondrial fragmentation in Prkaa2-deficient (prkaa2-/-) mice, suggesting that defective autophagy contributes to enhanced DNM1L expression and mitochondrial fragmentation. Additionally, the autophagic receptor protein SQSTM1/p62, which bound to DNM1L and led to its translocation into the autophagosome, was involved in DNM1L degradation by the autophagy-lysosome pathway. Gene silencing of SQSTM1 markedly reduced the association between SQSTM1 and DNM1L, impaired the degradation of DNM1L, and enhanced mitochondrial fragmentation in PRKAA-deficient endothelial cells. Finally, the genetic (DNM1L siRNA) or pharmacological (mdivi-1) inhibition of DNMA1L ablated mitochondrial fragmentation in the mouse aortic endothelium and prevented the acetylcholine-induced relaxation of isolated mouse aortas. This suggests that aberrant DNM1L is responsible for enhanced mitochondrial fragmentation and endothelial dysfunction in prkaa knockout mice. Overall, our results show that PRKAA deletion promoted mitochondrial fragmentation in vascular endothelial cells by inhibiting the autophagy-dependent degradation of DNM1L.
Keywords: AMPK; DNM1L; PRKAA/AMPK catalytic subunit α; autophagy; endothelial dysfunction; mitochondrial fission.
Figures
References
-
- Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta 2008; 1777:1092-7; PMID:18519024; http://dx.doi.org/ 10.1016/j.bbabio.2008.05.001 - DOI - PMC - PubMed
-
- Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev 2009; 89:799-845; PMID:19584314; http://dx.doi.org/ 10.1152/physrev.00030.2008 - DOI - PubMed
-
- Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A 2006; 103:2653-8; PMID:16477035; http://dx.doi.org/ 10.1073/pnas.0511154103 - DOI - PMC - PubMed
-
- Burte F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 2015; 11:11-24; PMID:25486875; http://dx.doi.org/ 10.1038/nrneurol.2014.228 - DOI - PubMed
-
- Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, et al.. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011; 124:444-53; PMID:21747057; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.110.014506 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous