

Impact of disease-Linked mutations targeting the oligomerization interfaces of aldehyde dehydrogenase 7A1
- PMID: 28087462
- PMCID: PMC5503811
- DOI: 10.1016/j.cbi.2017.01.002
Impact of disease-Linked mutations targeting the oligomerization interfaces of aldehyde dehydrogenase 7A1
Abstract
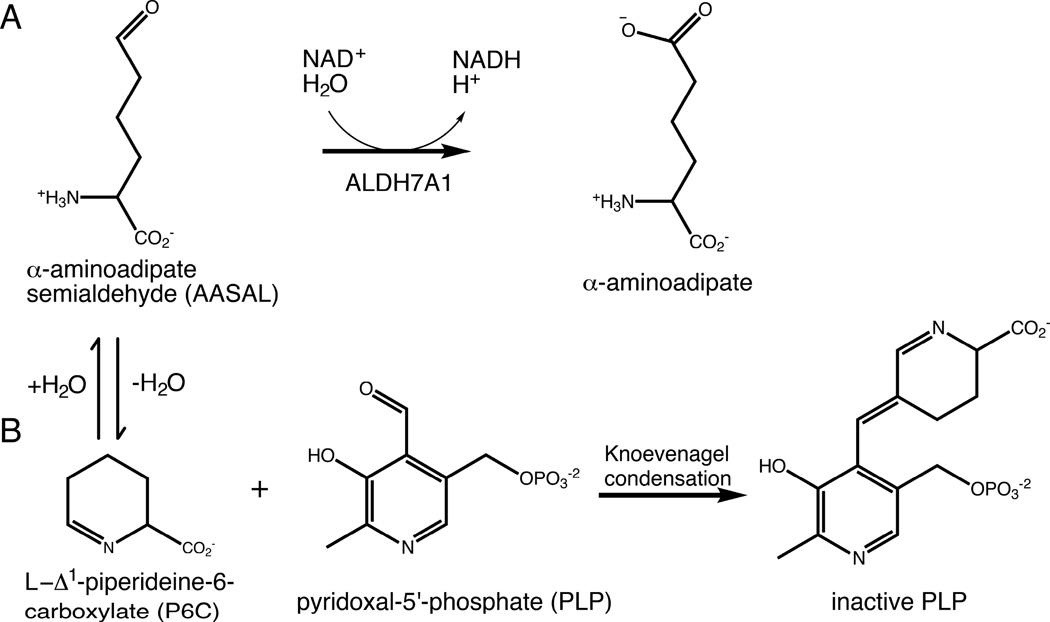
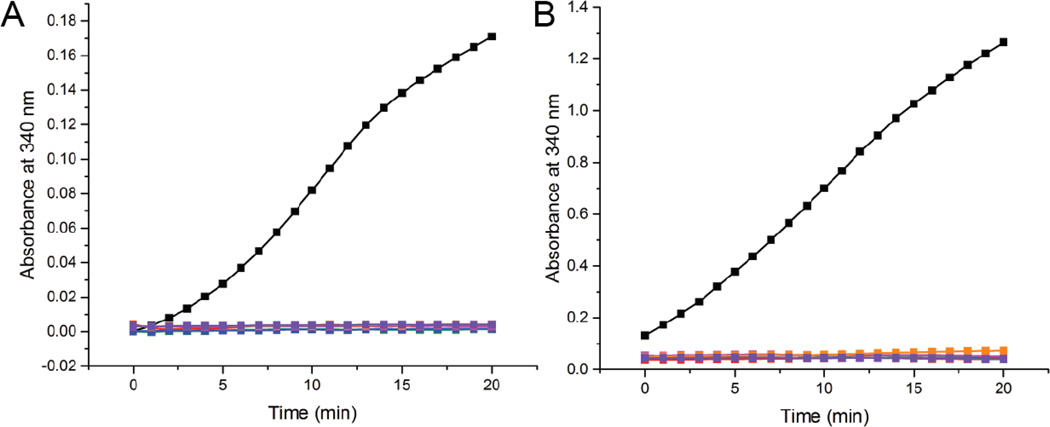
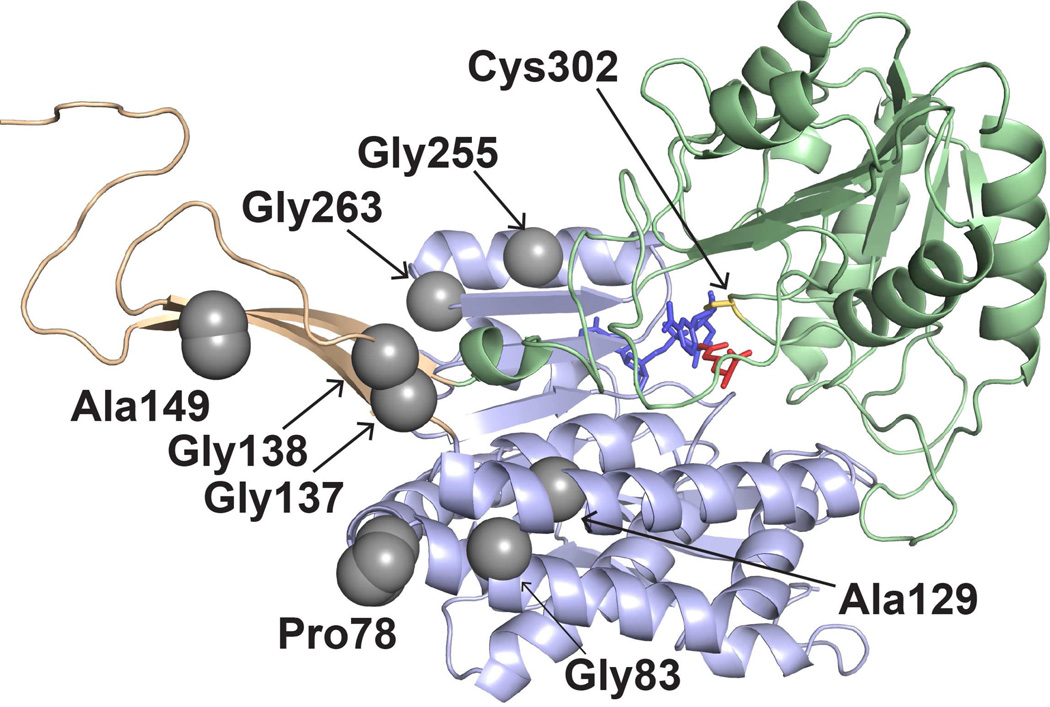
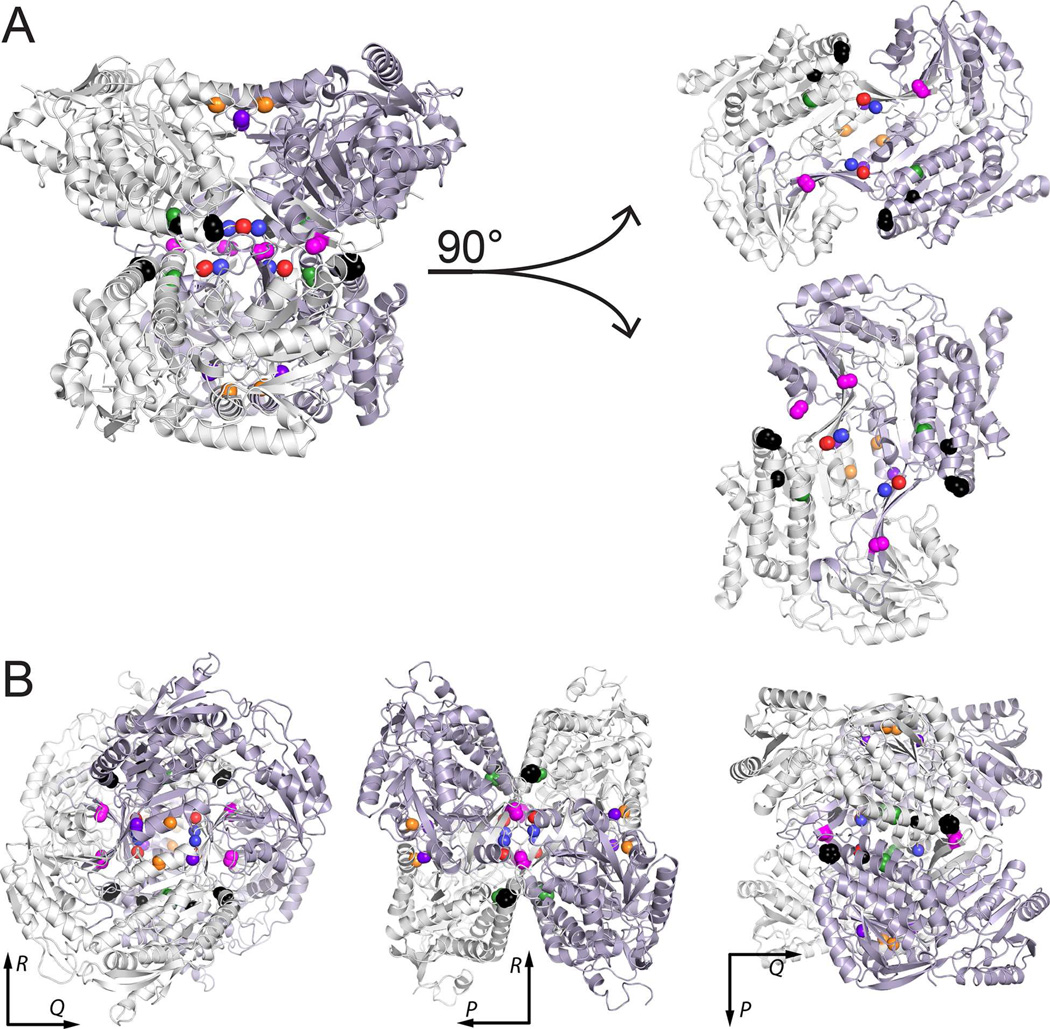
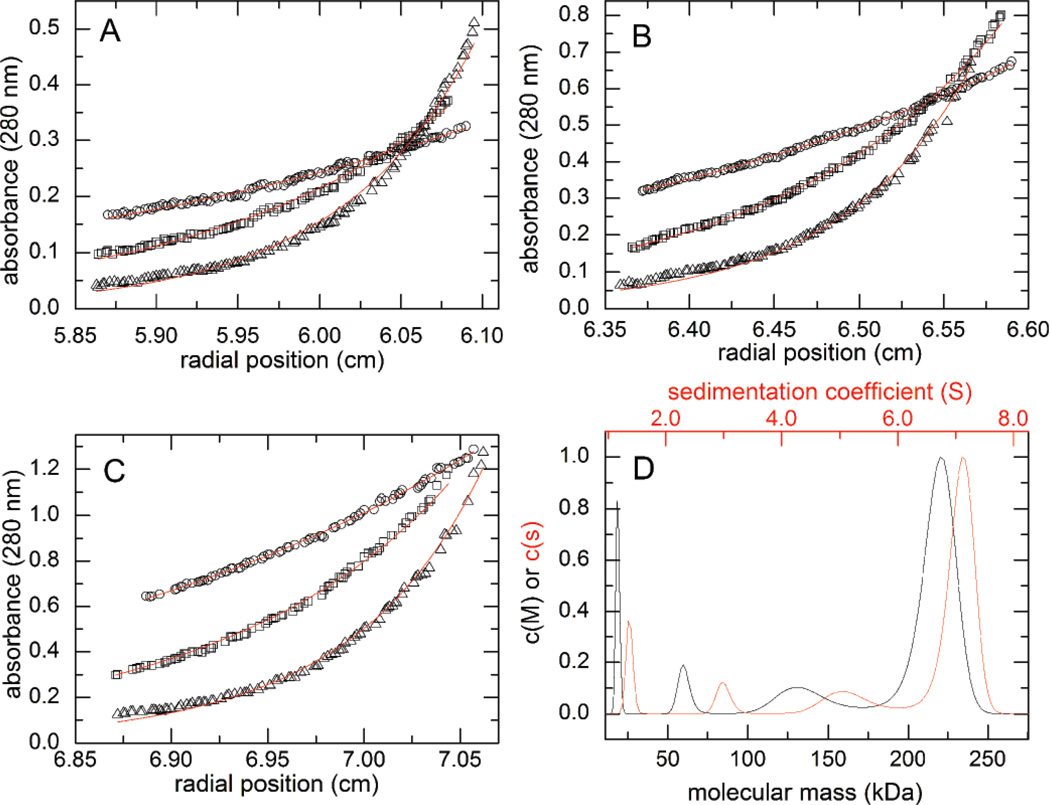
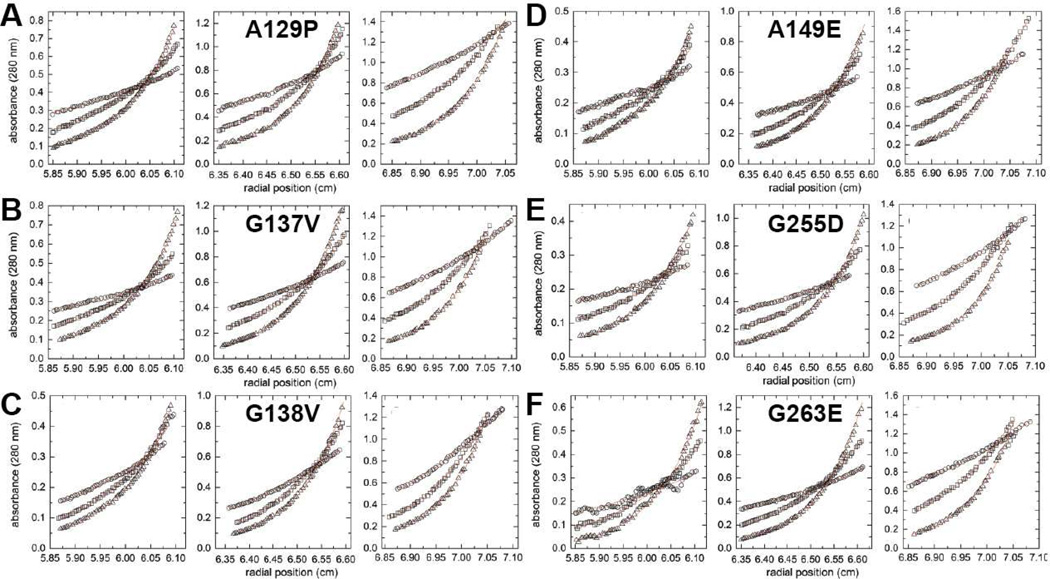
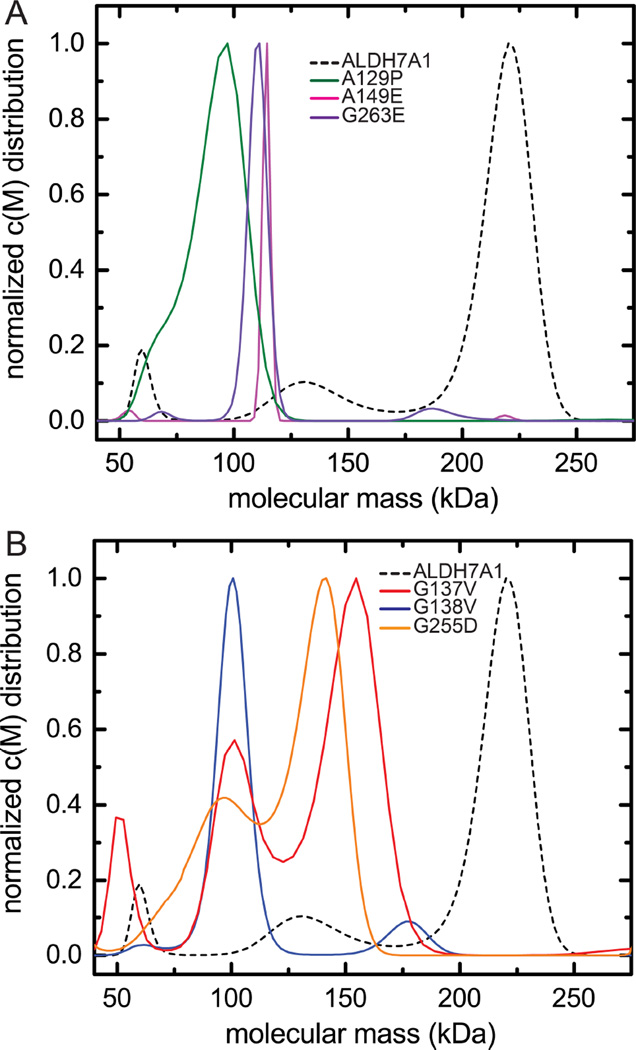
Aldehyde dehydrogenase 7A1 (ALDH7A1) is involved in lysine catabolism, catalyzing the oxidation of α-aminoadipate semialdehyde to α-aminoadipate. Certain mutations in the ALDH7A1 gene, which are presumed to reduce catalytic activity, cause an autosomal recessive seizure disorder known as pyridoxine-dependent epilepsy (PDE). Although the genetic association between ALDH7A1 and PDE is well established, little is known about the impact of PDE-mutations on the structure and catalytic function of the enzyme. Herein we report the first study of the molecular consequences of PDE mutations using purified ALDH7A1 variants. Eight variants, with mutations in the oligomer interfaces, were expressed in Escherichia coli: P78L, G83E, A129P, G137V, G138V, A149E, G255D, and G263E. All but P78L and G83E were soluble and could be purified. All six soluble mutants were catalytically inactive. The impact of the mutations on oligomerization was assessed by analytical ultracentrifugation. Wild-type ALDH7A1 is shown to exist in a dimer-tetramer equilibrium with a dissociation constant of 16 μM. In contrast to the wild-type enzyme, the variants reside in monomer-dimer equilibria and are apparently incapable of forming a tetrameric species, even at high enzyme concentration. The available evidence suggests that they are misfolded assemblies lacking the three-dimensional structure required for catalysis.
Keywords: ALDH7A1; Aldehyde dehydrogenase; Analytical ultracentrifugation; Lysine catabolism; Protein oligomerization; Pyridoxine-dependent epilepsy.
Copyright © 2017 Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Ma I, Allan AL. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem. Cell. Rev. 2011;7:292–306. - PubMed
-
- Muzio G, Maggiora M, Paiuzzi E, Oraldi M, Canuto RA. Aldehyde dehydrogenases and cell proliferation. Free Radic. Biol. Med. 2012;52:735–746. - PubMed
-
- Mills PB, Struys E, Jakobs C, Plecko B, Baxter P, Baumgartner M, Willemsen MA, Omran H, Tacke U, Uhlenberg B, Weschke B, Clayton PT. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat. Med. 2006;12:307–309. - PubMed
-
- Stockler S, Plecko B, Gospe SM, Jr, Coulter-Mackie M, Connolly M, van Karnebeek C, Mercimek-Mahmutoglu S, Hartmann H, Scharer G, Struijs E, Tein I, Jakobs C, Clayton P, Van Hove JL. Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol. Genet. Metab. 2011;104:48–60. - PubMed
-
- Stenson PD, Ball E, Howells K, Phillips A, Mort M, Cooper DN. Human Gene Mutation Database: towards a comprehensive central mutation database. J Med Genet. 2008;45:124–126. - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
