

Calmodulin limits pathogenic Na+ channel persistent current
- PMID: 28087622
- PMCID: PMC5299624
- DOI: 10.1085/jgp.201611721
Calmodulin limits pathogenic Na+ channel persistent current
Abstract
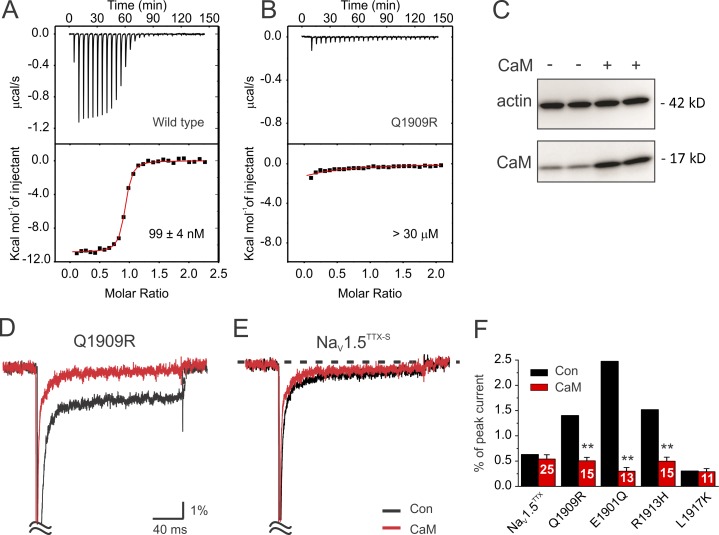
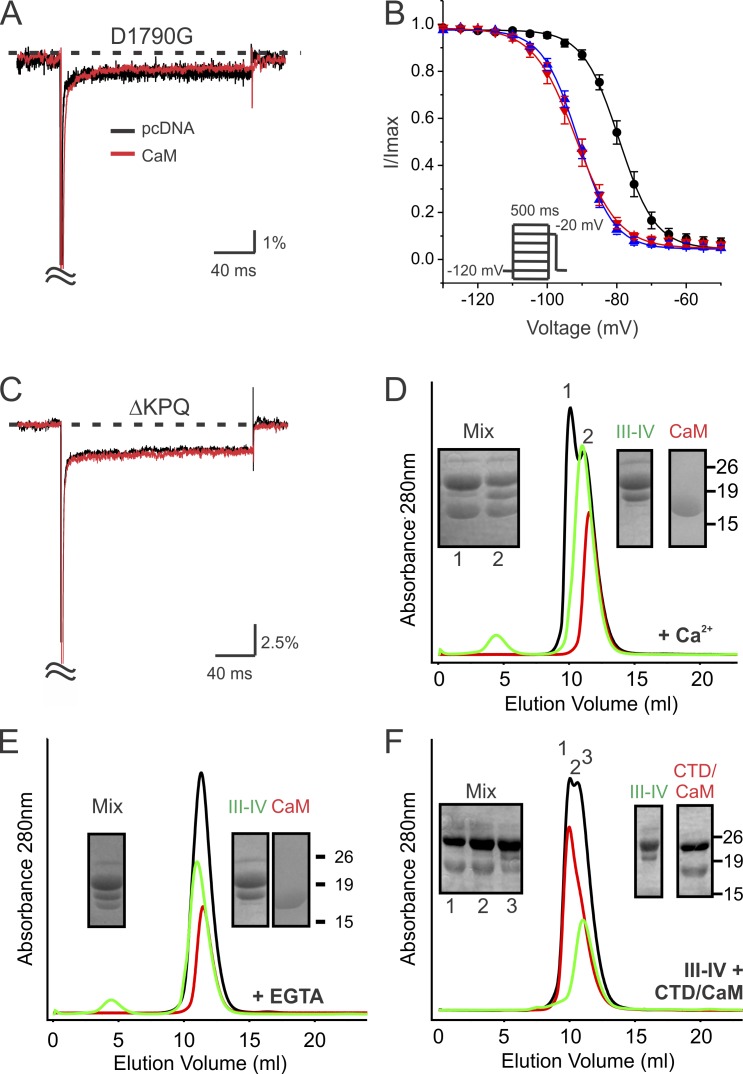
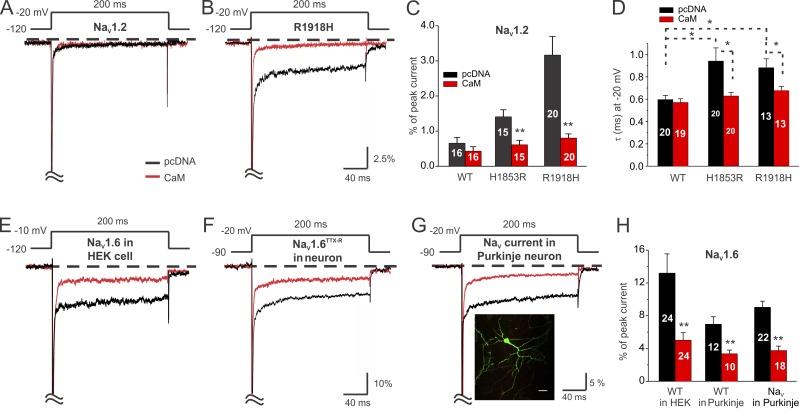
Increased "persistent" current, caused by delayed inactivation, through voltage-gated Na+ (NaV) channels leads to cardiac arrhythmias or epilepsy. The underlying molecular contributors to these inactivation defects are poorly understood. Here, we show that calmodulin (CaM) binding to multiple sites within NaV channel intracellular C-terminal domains (CTDs) limits persistent Na+ current and accelerates inactivation across the NaV family. Arrhythmia or epilepsy mutations located in NaV1.5 or NaV1.2 channel CTDs, respectively, reduce CaM binding either directly or by interfering with CTD-CTD interchannel interactions. Boosting the availability of CaM, thus shifting its binding equilibrium, restores wild-type (WT)-like inactivation in mutant NaV1.5 and NaV1.2 channels and likewise diminishes the comparatively large persistent Na+ current through WT NaV1.6, whose CTD displays relatively low CaM affinity. In cerebellar Purkinje neurons, in which NaV1.6 promotes a large physiological persistent Na+ current, increased CaM diminishes the persistent Na+ current, suggesting that the endogenous, comparatively weak affinity of NaV1.6 for apoCaM is important for physiological persistent current.
© 2017 Yan et al.
Figures


References
-
- Benhorin J., Goldmit M., MacCluer J.W., Blangero J., Goffen R., Leibovitch A., Rahat A., Wang Q., Medina A., Towbin J., and Kerem B.. 1998. Identification of a new SCN5A mutation, D1840G, associated with the long QT syndrome. Hum. Mutat. 12:72 10.1002/(SICI)1098-1004(1998)12:1<72::AID-HUMU17>3.0.CO;2-Z - DOI - PubMed
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
