

Mitochondrial redox plays a critical role in the paradoxical effects of NAPDH oxidase-derived ROS on coronary endothelium
- PMID: 28088753
- PMCID: PMC5340144
- DOI: 10.1093/cvr/cvw249
Mitochondrial redox plays a critical role in the paradoxical effects of NAPDH oxidase-derived ROS on coronary endothelium
Abstract
Aims: There are conflicting reports on the role of reactive oxygen species (ROS) i.e. beneficial vs. harmful, in vascular endothelium. Here, we aim to examine whether duration of exposure to ROS and/or subcellular ROS levels are responsible for the apparently paradoxical effects of oxidants on endothelium.
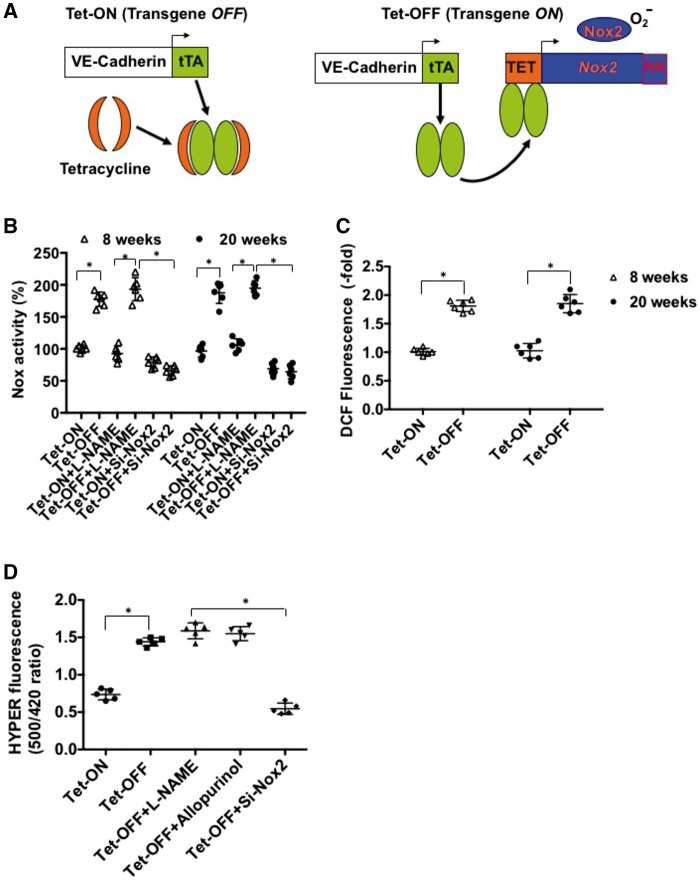
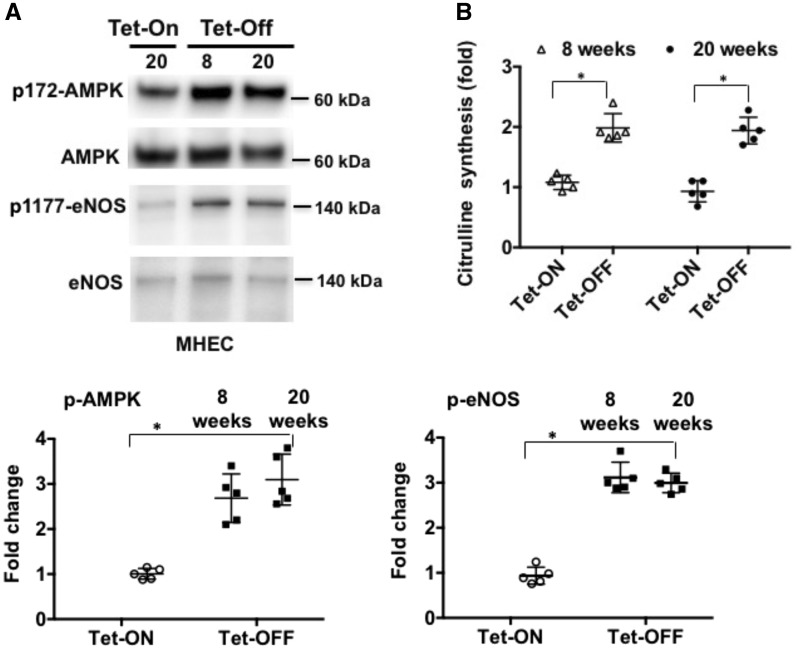
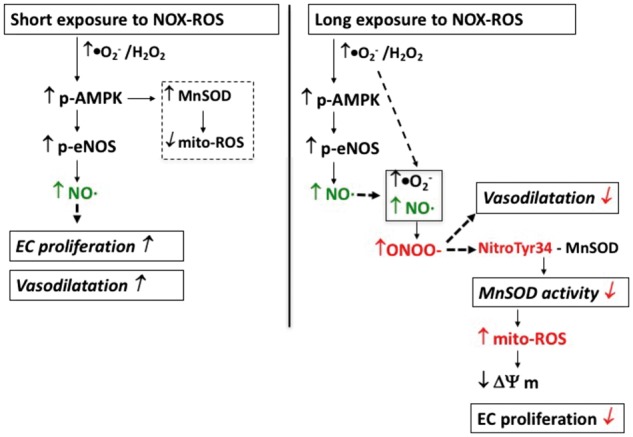
Methods and results: We have recently generated binary (Tet-ON/OFF) conditional transgenic mice (Tet-Nox2:VE-Cad-tTA) that can induce 1.8 ± 0.42-fold increase in NADPH oxidase (NOX)-derived ROS specifically in vascular endothelium upon withdrawal of tetracycline from the drinking water. Animals were divided in two groups: one exposed to high endogenous ROS levels for 8 weeks (short-term) and the other for 20 weeks (long-term). Using endothelial cells (EC) isolated from mouse hearts (MHEC), we demonstrate that both short-term and long-term increase in NOX-ROS induced AMPK-mediated activation of eNOS. Interestingly, although endothelium-dependent nitric oxide (NO)-mediated coronary vasodilation was significantly increased after short-term increase in NOX-ROS, coronary vasodilation was drastically reduced after long-term increase in ROS. We also show that short-term ROS increase induced proliferation in EC and angiogenic sprouting in the aorta. In contrast, long-term increase in cytosolic ROS resulted in nitrotyrosine-mediated inactivation of mitochondrial (mito) antioxidant MnSOD, increase in mito-ROS, loss of mitochondrial membrane potential (Δψm), decreased EC proliferation and angiogenesis.
Conclusion: The findings suggest that NOX-derived ROS results in increased mito-ROS. Whereas short-term increase in mito-ROS was counteracted by MnSOD, long-term increase in ROS resulted in nitrotyrosine-mediated inactivation of MnSOD, leading to unchecked increase in mito-ROS and loss of Δψm followed by inhibition of endothelial function and proliferation.
Keywords: Endothelium • Signal transduction • Nitric oxide • Reactive oxygen species • NADPH oxidase.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author 2017. For Permissions, please email: journals.permissions@oup.com.
Figures




References
-
- Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol 2003;91:7A–11A. - PubMed
-
- Irani K. Oxidant signaling in vascular cell growth, death, and survival : a review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circ Res 2000;87:179–183. - PubMed
-
- Abid MR, Tsai JC, Spokes KC, Deshpande SS, Irani K, Aird WC. Vascular endothelial growth factor induces manganese-superoxide dismutase expression in endothelial cells by a Rac1-regulated NADPH oxidase-dependent mechanism. FASEB J 2001;15:2548–2550. - PubMed
-
- Moldovan L, Irani K, Moldovan NI, Finkel T, Goldschmidt-Clermont PJ. The actin cytoskeleton reorganization induced by Rac1 requires the production of superoxide. Antioxid Redox Signal 1999;1:29–43. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
