

Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R
- PMID: 28088763
- PMCID: PMC5408845
- DOI: 10.1093/bioinformatics/btw777
Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R
Abstract
Motivation: Single-cell RNA sequencing (scRNA-seq) is increasingly used to study gene expression at the level of individual cells. However, preparing raw sequence data for further analysis is not a straightforward process. Biases, artifacts and other sources of unwanted variation are present in the data, requiring substantial time and effort to be spent on pre-processing, quality control (QC) and normalization.
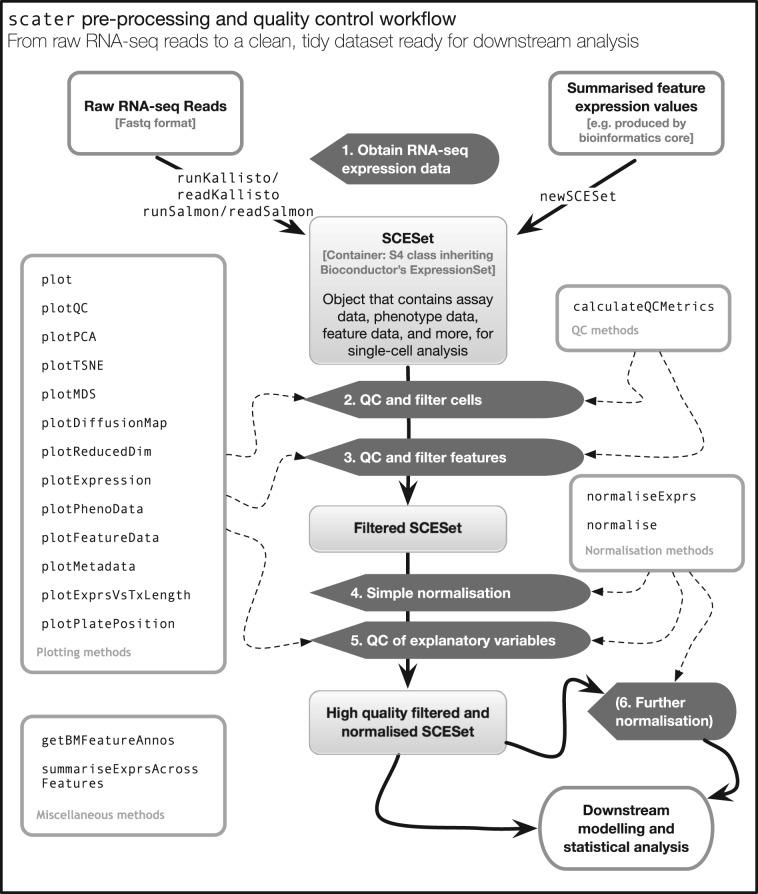
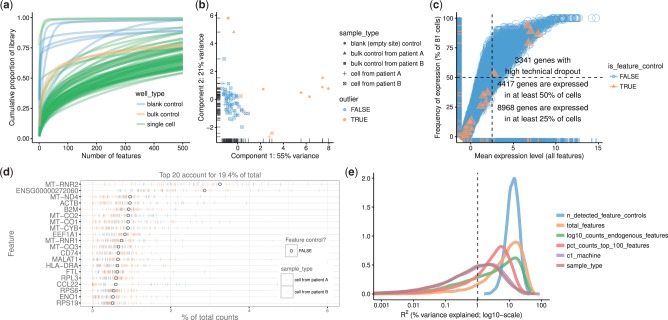
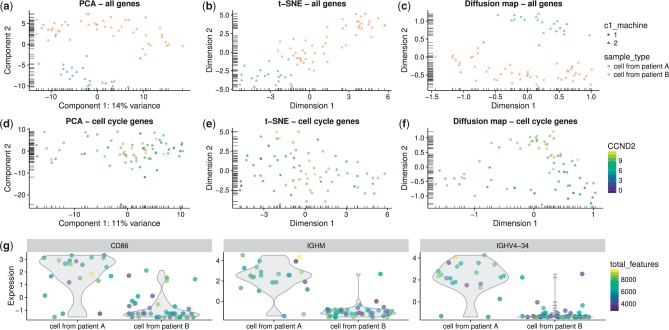
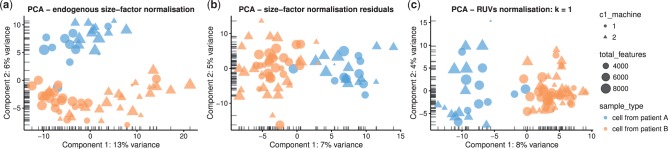
Results: We have developed the R/Bioconductor package scater to facilitate rigorous pre-processing, quality control, normalization and visualization of scRNA-seq data. The package provides a convenient, flexible workflow to process raw sequencing reads into a high-quality expression dataset ready for downstream analysis. scater provides a rich suite of plotting tools for single-cell data and a flexible data structure that is compatible with existing tools and can be used as infrastructure for future software development.
Availability and implementation: The open-source code, along with installation instructions, vignettes and case studies, is available through Bioconductor at http://bioconductor.org/packages/scater .
Contact: davis@ebi.ac.uk.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author 2017. Published by Oxford University Press.
Figures
References
-
- Angerer P. et al. (2015) destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics, 32, 1241–1243. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
