

Pulmonary vascular and ventricular dysfunction in the susceptible patient (2015 Grover Conference series)
- PMID: 28090285
- PMCID: PMC5210067
- DOI: 10.1086/688315
Pulmonary vascular and ventricular dysfunction in the susceptible patient (2015 Grover Conference series)
Erratum in
-
Corrigendum.Pulm Circ. 2017 Apr-Jun;7(2):559. doi: 10.1177/2045893217706334. Pulm Circ. 2017. PMID: 28597768 Free PMC article. No abstract available.
Abstract
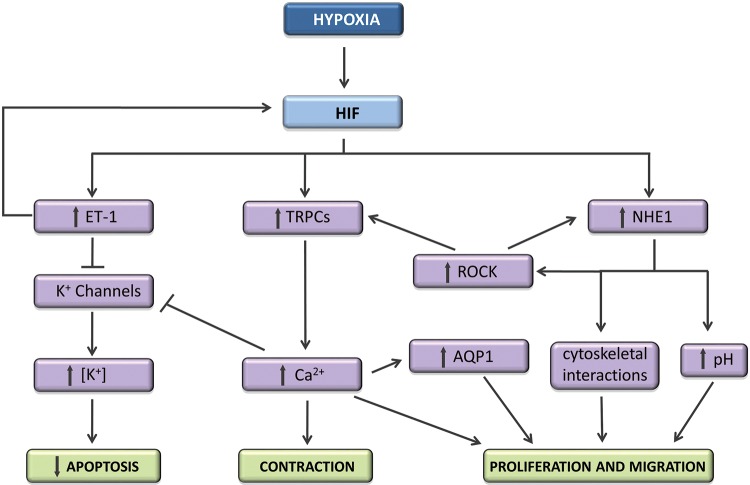
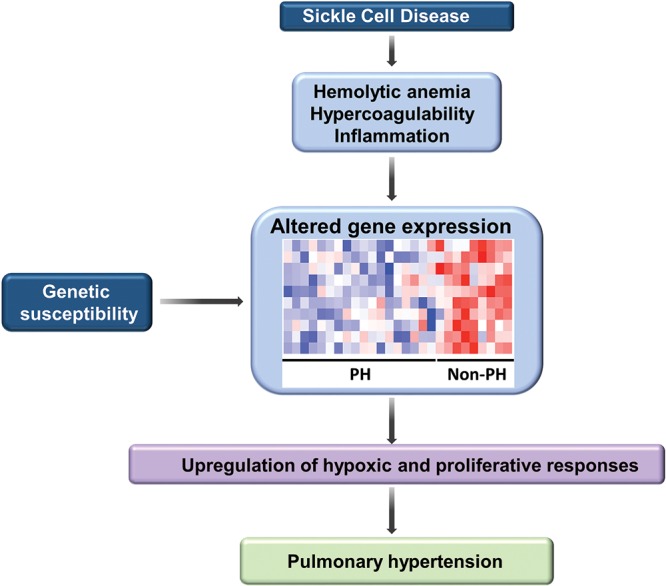
Pulmonary blood vessel structure and tone are maintained by a complex interplay between endogenous vasoactive factors and oxygen-sensing intermediaries. Under physiological conditions, these signaling networks function as an adaptive interface between the pulmonary circulation and environmental or acquired perturbations to preserve oxygenation and maintain systemic delivery of oxygen-rich hemoglobin. Chronic exposure to hypoxia, however, triggers a range of pathogenetic mechanisms that include hypoxia-inducible factor 1α (HIF-1α)-dependent upregulation of the vasoconstrictor peptide endothelin 1 in pulmonary endothelial cells. In pulmonary arterial smooth muscle cells, chronic hypoxia induces HIF-1α-mediated upregulation of canonical transient receptor potential proteins, as well as increased Rho kinase-Ca2+ signaling and pulmonary arteriole synthesis of the profibrotic hormone aldosterone. Collectively, these mechanisms contribute to a contractile or hypertrophic pulmonary vascular phenotype. Genetically inherited disorders in hemoglobin structure are also an important etiology of abnormal pulmonary vasoreactivity. In sickle cell anemia, for example, consumption of the vasodilator and antimitogenic molecule nitric oxide by cell-free hemoglobin is an important mechanism underpinning pulmonary hypertension. Contemporary genomic and transcriptomic analytic methods have also allowed for the discovery of novel risk factors relevant to sickle cell disease, including GALNT13 gene variants. In this report, we review cutting-edge observations characterizing these and other pathobiological mechanisms that contribute to pulmonary vascular and right ventricular vulnerability.
Keywords: genetics; hypoxia; pulmonary hypertension.
Figures

References
-
- Anand IS, Wu T. Syndromes of subacute mountain sickness. High Alt Med Biol 2004;5(2):156–170. - PubMed
-
- Singh I, Kapila CC, Khanna PK, Nanda RB, Rao BD. High-altitude pulmonary oedema. Lancet 1965;285(7379):229–234. - PubMed
-
- Menon ND. High-altitude pulmonary edema: a clinical study. N Engl J Med 1965;273(2):66–73. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
