

Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating
- PMID: 28092267
- PMCID: PMC5344670
- DOI: 10.7554/eLife.24149
Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating
Abstract
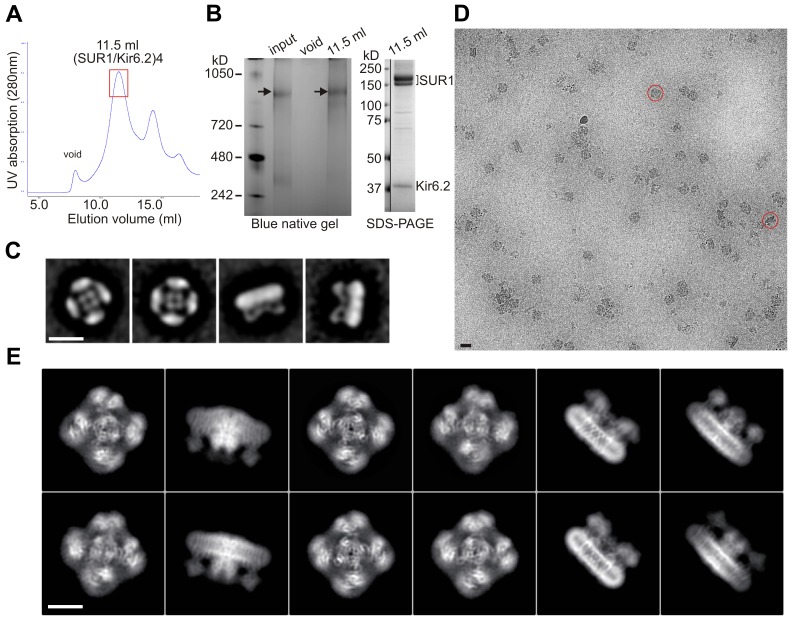
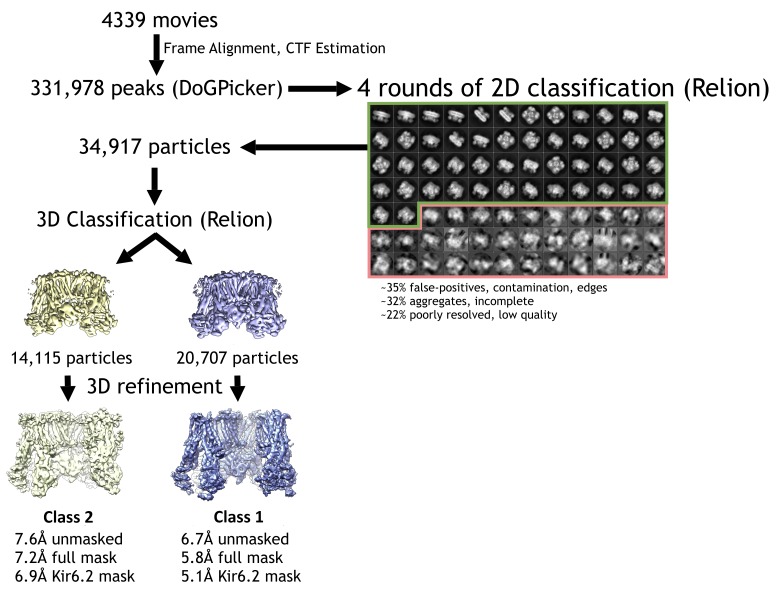
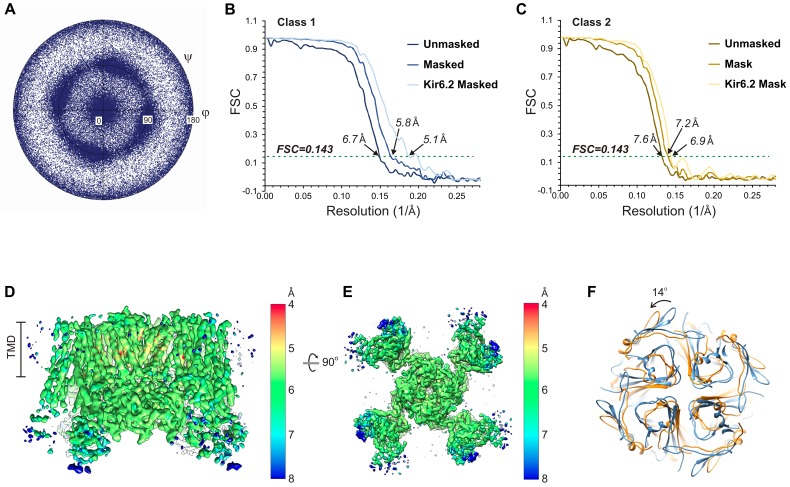
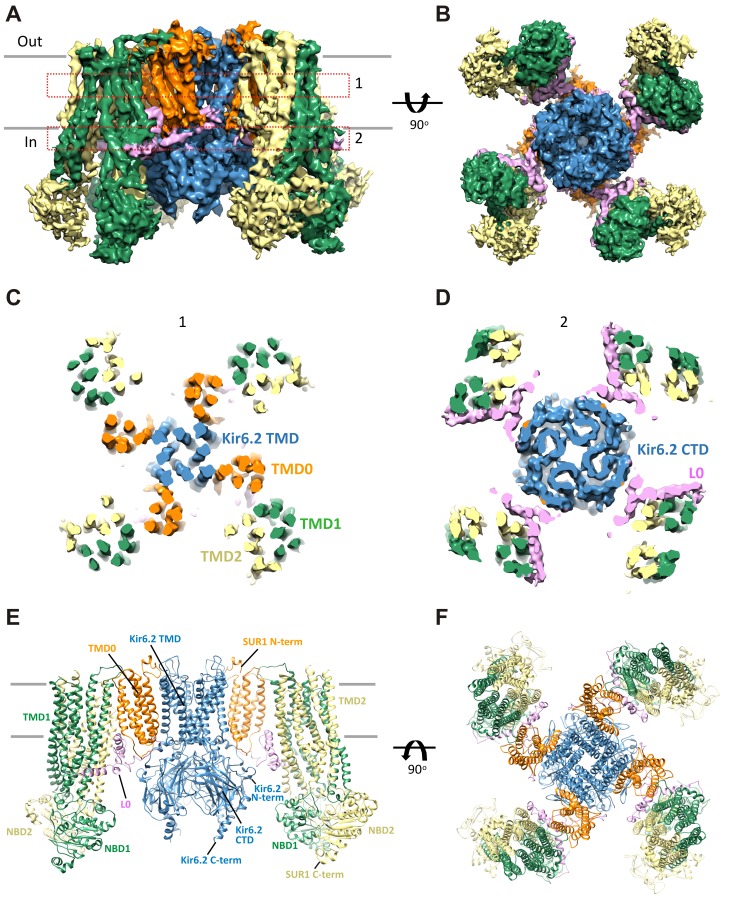
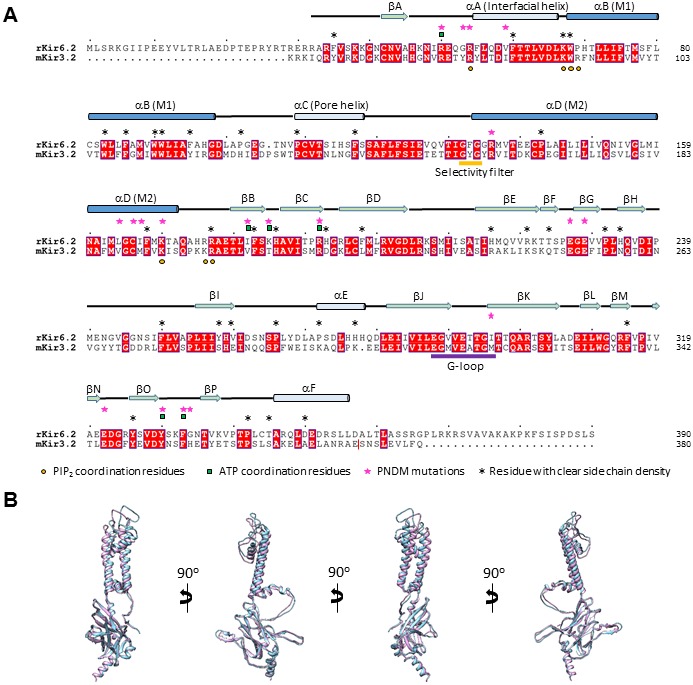
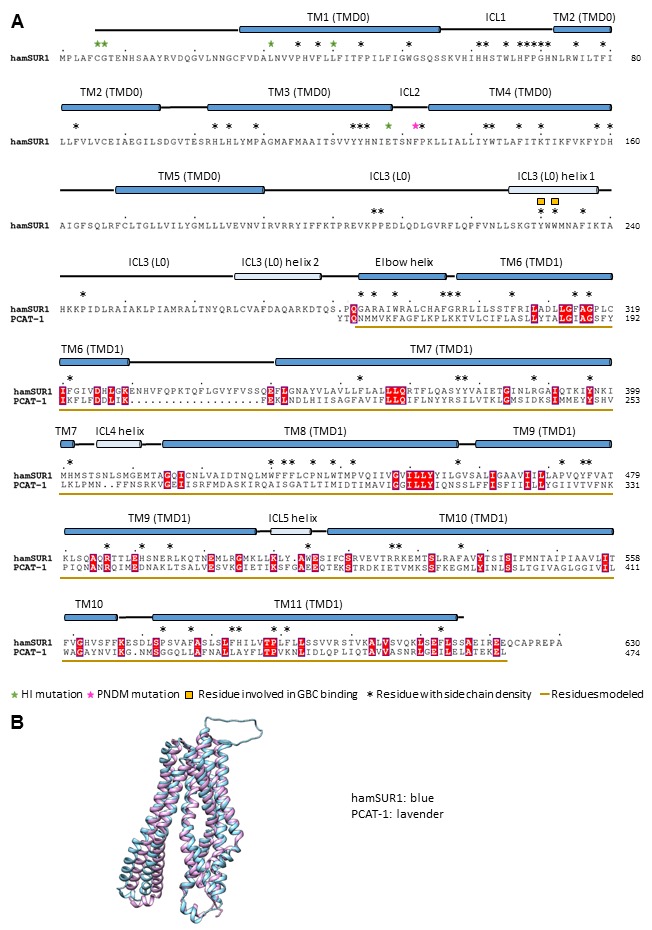
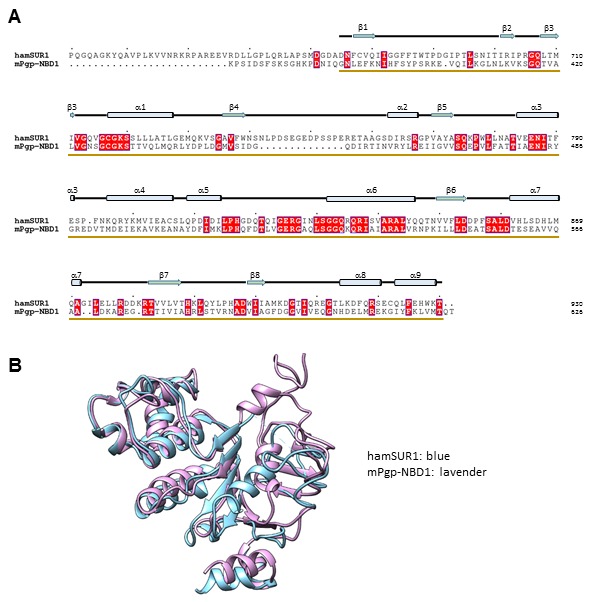
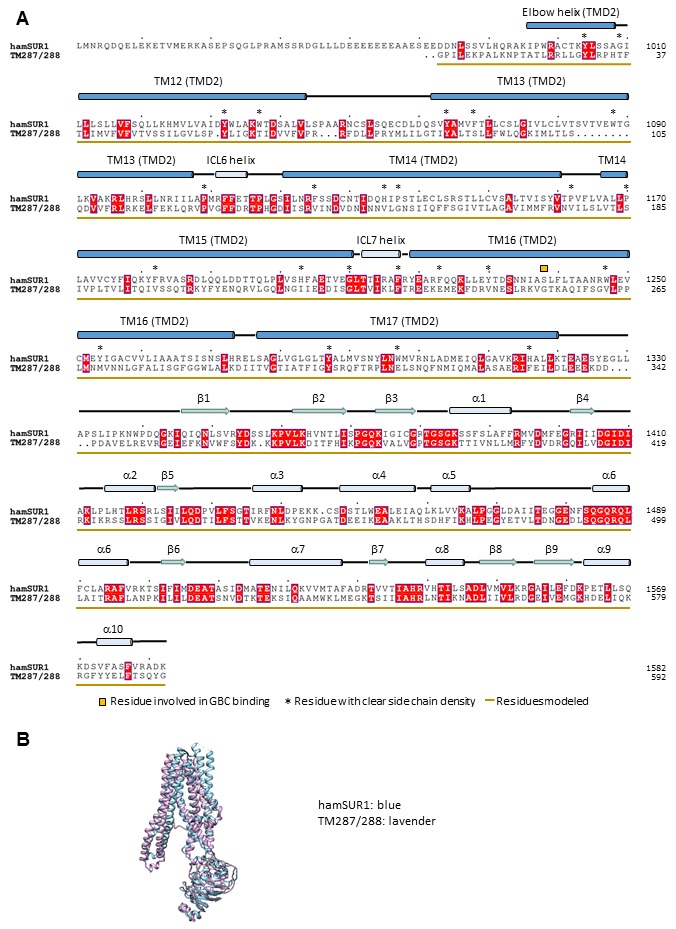
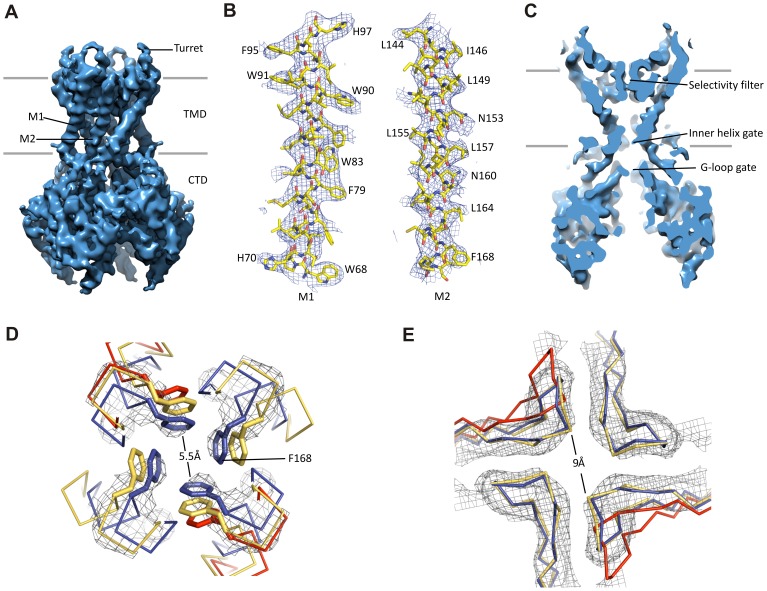
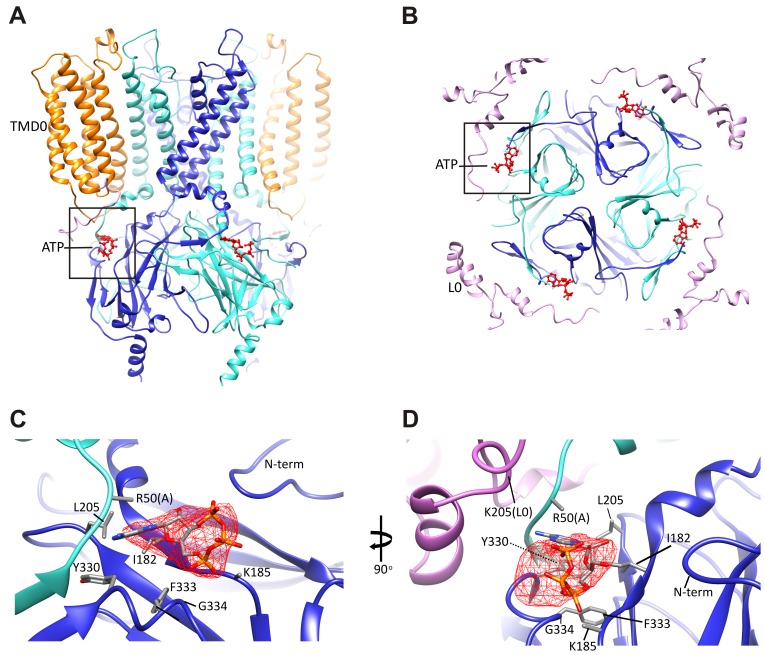
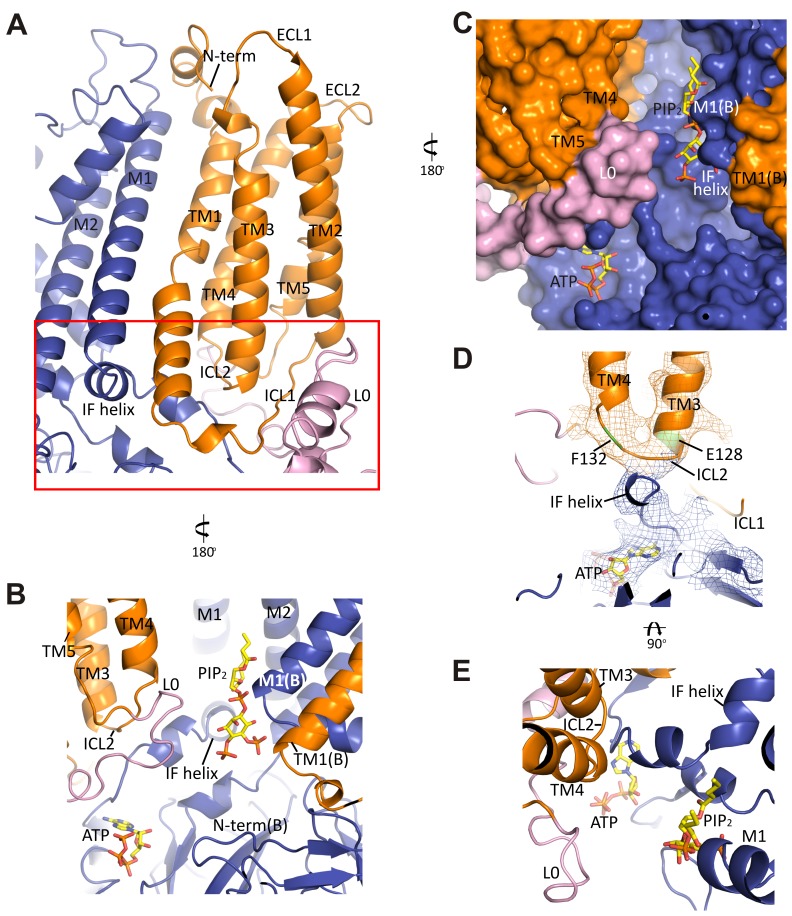
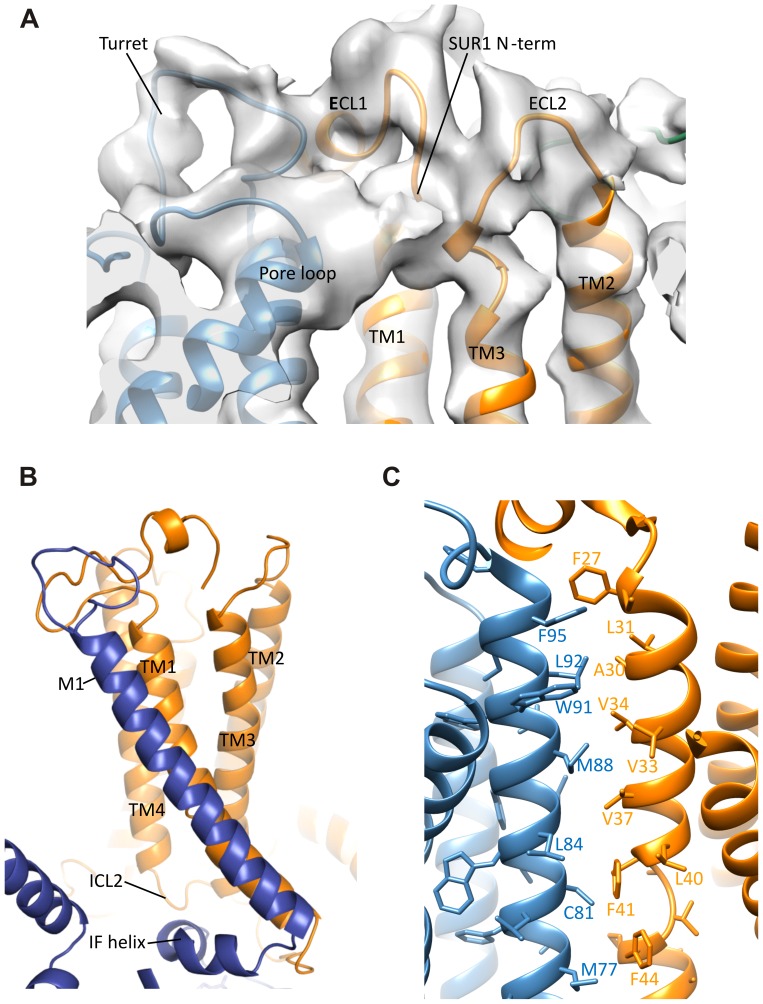
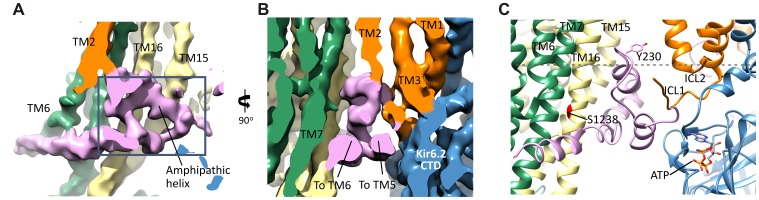
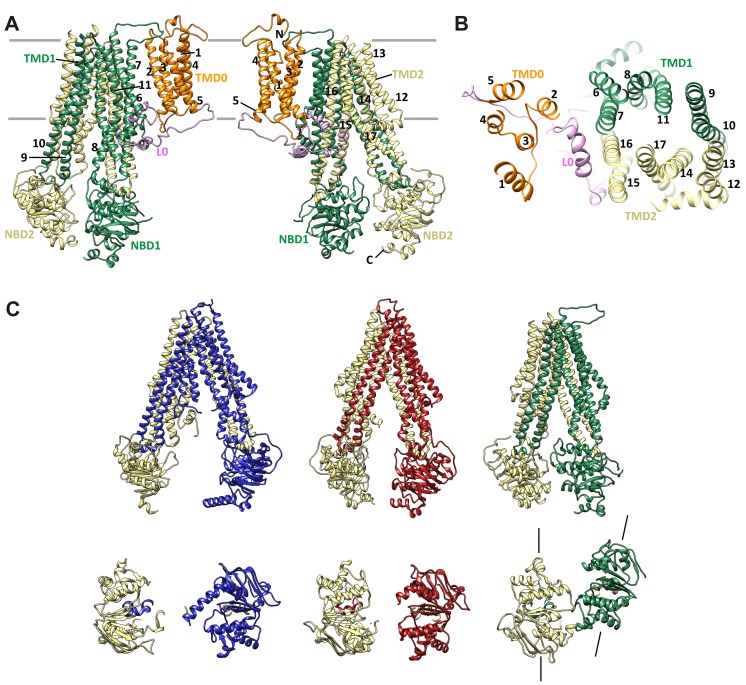
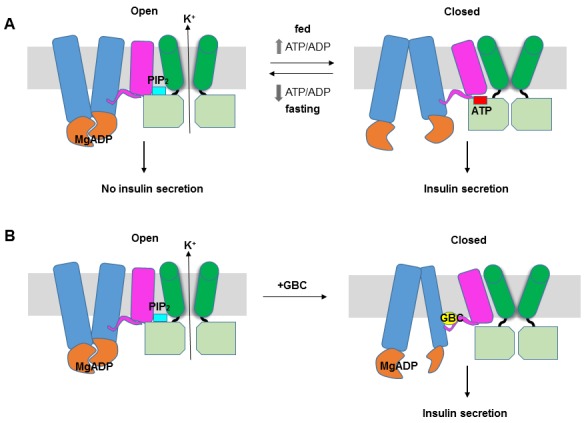
KATP channels are metabolic sensors that couple cell energetics to membrane excitability. In pancreatic β-cells, channels formed by SUR1 and Kir6.2 regulate insulin secretion and are the targets of antidiabetic sulfonylureas. Here, we used cryo-EM to elucidate structural basis of channel assembly and gating. The structure, determined in the presence of ATP and the sulfonylurea glibenclamide, at ~6 Å resolution reveals a closed Kir6.2 tetrameric core with four peripheral SUR1s each anchored to a Kir6.2 by its N-terminal transmembrane domain (TMD0). Intricate interactions between TMD0, the loop following TMD0, and Kir6.2 near the proposed PIP2 binding site, and where ATP density is observed, suggest SUR1 may contribute to ATP and PIP2 binding to enhance Kir6.2 sensitivity to both. The SUR1-ABC core is found in an unusual inward-facing conformation whereby the two nucleotide binding domains are misaligned along a two-fold symmetry axis, revealing a possible mechanism by which glibenclamide inhibits channel activity.
Keywords: ABC transporter; ATP; biophysics; inward rectifier; none; structural biology; sulfonylurea; sulfonylurea receptor.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures
Comment in
-
From ions to insulin.Elife. 2017 Mar 9;6:e25159. doi: 10.7554/eLife.25159. Elife. 2017. PMID: 28276322 Free PMC article.
References
-
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D Biological Crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
