

Genomewide landscape of gene-metabolome associations in Escherichia coli
- PMID: 28093455
- PMCID: PMC5293155
- DOI: 10.15252/msb.20167150
Genomewide landscape of gene-metabolome associations in Escherichia coli
Abstract
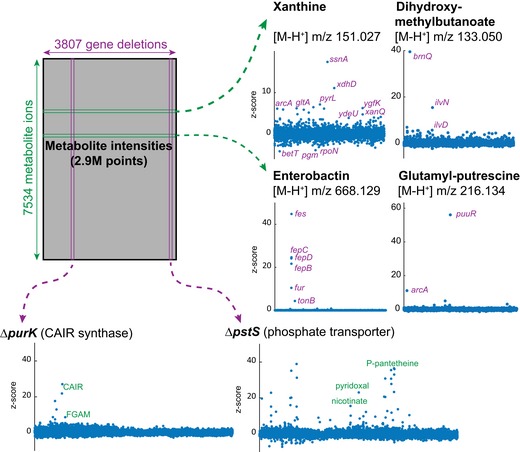
Metabolism is one of the best-understood cellular processes whose network topology of enzymatic reactions is determined by an organism's genome. The influence of genes on metabolite levels, however, remains largely unknown, particularly for the many genes encoding non-enzymatic proteins. Serendipitously, genomewide association studies explore the relationship between genetic variants and metabolite levels, but a comprehensive interaction network has remained elusive even for the simplest single-celled organisms. Here, we systematically mapped the association between > 3,800 single-gene deletions in the bacterium Escherichia coli and relative concentrations of > 7,000 intracellular metabolite ions. Beyond expected metabolic changes in the proximity to abolished enzyme activities, the association map reveals a largely unknown landscape of gene-metabolite interactions that are not represented in metabolic models. Therefore, the map provides a unique resource for assessing the genetic basis of metabolic changes and conversely hypothesizing metabolic consequences of genetic alterations. We illustrate this by predicting metabolism-related functions of 72 so far not annotated genes and by identifying key genes mediating the cellular response to environmental perturbations.
Keywords: GWAS; functional genomics; interaction network; metabolism; metabolomics.
© 2017 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
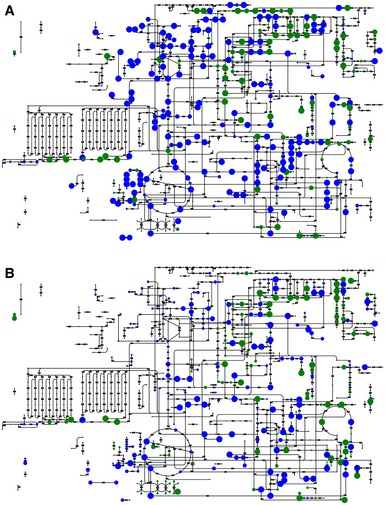
- A, B
Metabolites putatively detected in central metabolism for (A) negative mode and (B) positive mode ionization. Green circles represent compounds specifically associated with a single detected ion within 3 mDa tolerance. Blue circles represent compounds that are ambiguously associated with an ion, that is, compounds whose mass was found to match a detected ion but has not a unique molecular weight in the metabolic model used for annotation. The dot size is proportional to the annotation confidence. Additional compounds not depicted on the amended network were also putatively identified.
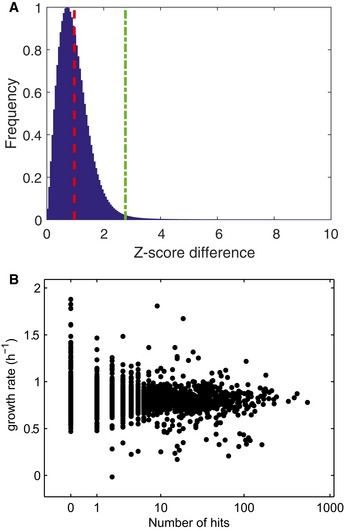
For each ion and deletion mutant, the absolute difference between Z‐scores of two biological replicates separately processed on different days was calculated. The overall distribution is shown in the histogram. Mean value (μ, red dashed line) and 99.9 confidence interval (μ + 3σ, green dashed line).
Growth‐rate dependence of metabolic changes. For each deletion mutant, the number of hits (largest 0.1% of metabolic changes) is plotted against the respective growth rate.
- A, B
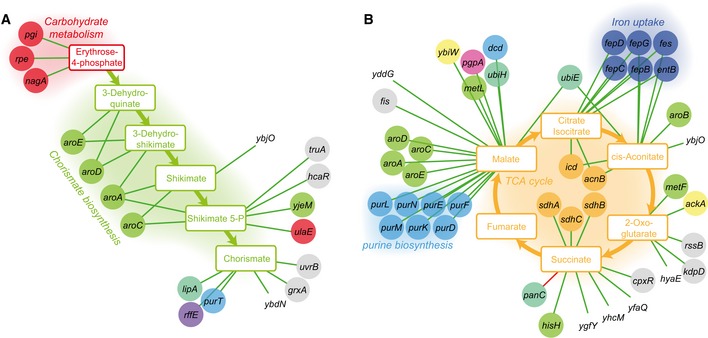
Genes associated with the detectable intermediates of chorismate metabolism (A) and the TCA cycle (B). To illustrate only high‐confidence links, we report only the edges associating a gene to deprotonated metabolites. Color code of metabolites and genes is as in Fig 2. Green and red edges represent increase and decrease in metabolite levels, respectively.
- A–C
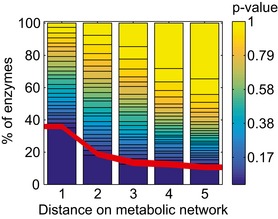
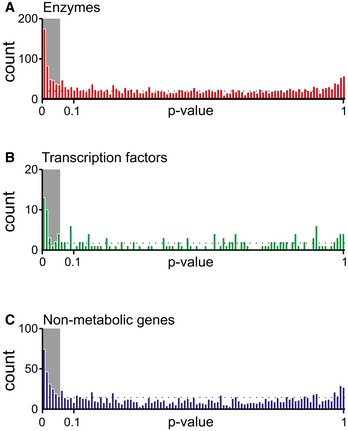
Locality analysis for genes encoding either enzymes, transcription factors, or protein of non‐metabolic function [e.g., ribosomes (Andres Leon et al, 2009)]. The distribution of the significance (i.e., P‐value, calculated from a permutation test) of the locality test for each of the tested genes is reported. Genes are grouped in three major classes: enzymes, transcription factors, and genes encoding for proteins that establish a physical interaction with at least one annotated enzyme (i.e., non‐metabolic genes). The gray region in the histogram highlights those gene deletions for which a significant local effect can be extrapolated (P‐value < 0.05). The overrepresentation of genes within this region supports a tendency for several knockouts to elicit local metabolic effects.
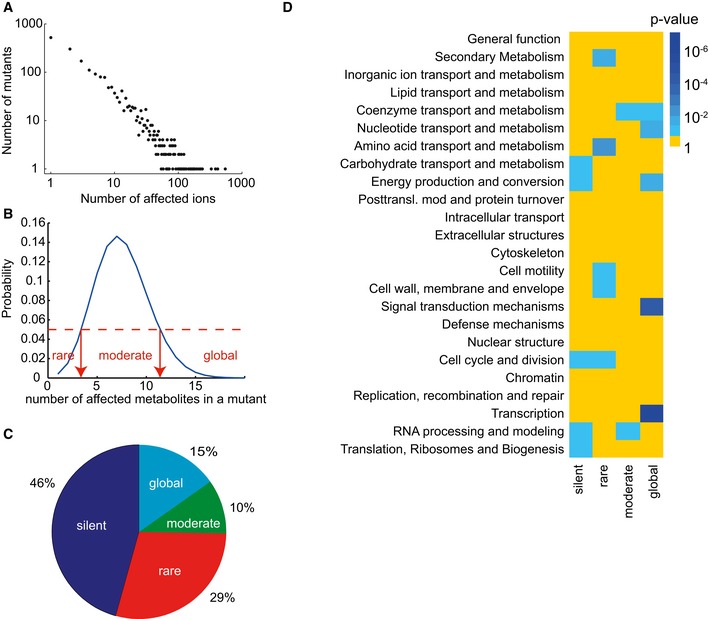
Frequency of metabolite changes in gene knockout mutants in the set of 0.1% most significant changes, seemingly following a power‐law distribution.
Definition of boundaries for the classification of mutants according to the number of differential ions present in the top 0.1% percentile.
Classification of mutants based on number of detectable changes in metabolome.
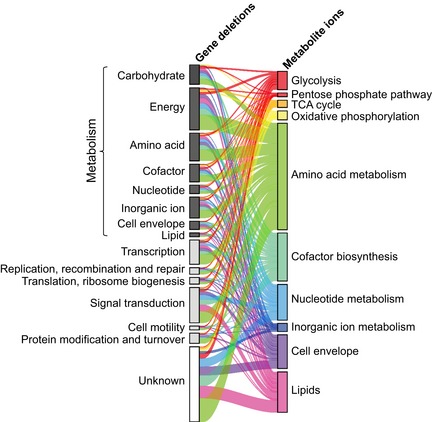
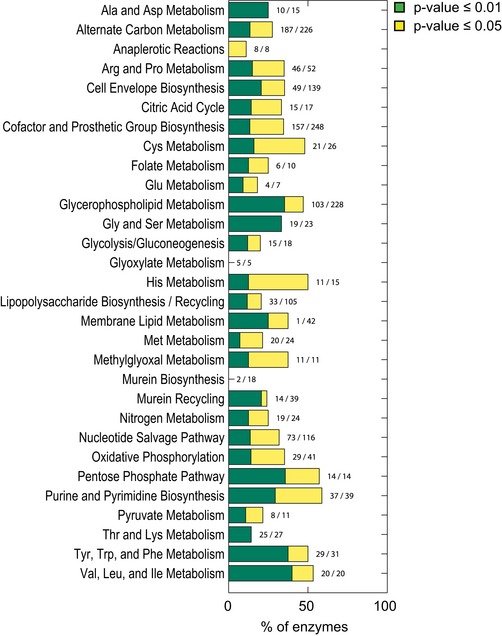
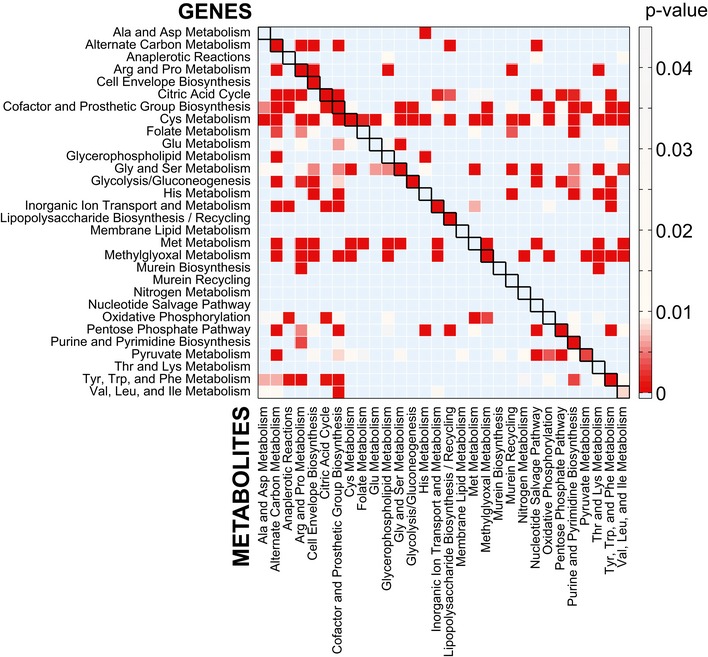
Cellular function enrichment analysis. Enrichment significance (P‐value) was derived by hypergeometric probability density function. Only significant enrichments with a P‐value < 0.1 are highlighted.
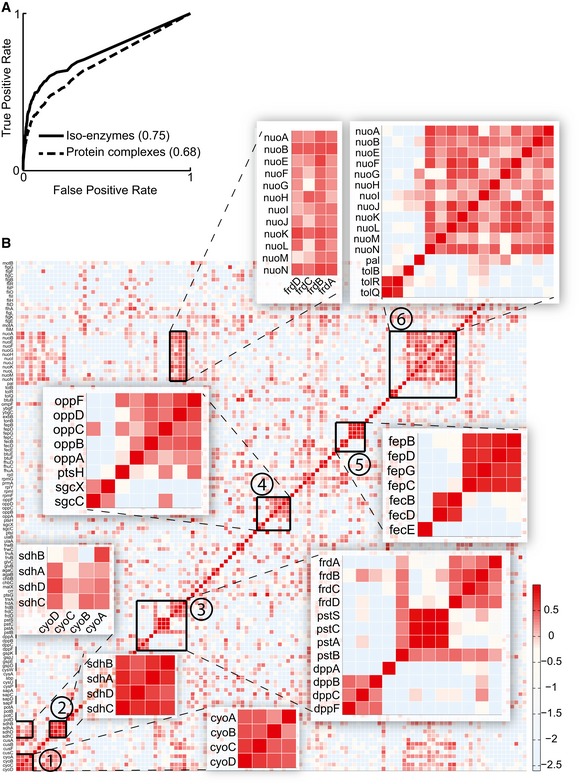
Recovery of enzyme function. Receiver operating characteristic curves obtained for the recovery of Escherichia coli isoenzymes and protein complexes based on the metabolome profiles recorded in single deletion mutants. The area under the curve (AUC) is reported in parentheses.
Consistent metabolic patterns in mutants of protein complex subunits. The heatmap shows the pair‐wise similarity (e.g., CLR index) between metabolome response to gene deletions. Genes related to densely connected protein complexes consisting of at least three subunits are selected. We visualized the protein complex adjacency matrix, opportunely reordered. Magnified protein complexes are 1, succinate dehydrogenase; 2, cytochrome bo terminal oxidase; 3, fumarate reductase/phosphate ABC transporter/dipeptide ABC transporter; 4, murein tripeptide ABC transporter; 5, ferric enterobactin transport complex/ferric dicitrate transport system; 6, NADH:ubiquinone oxidoreductase/Tol–Pal cell envelope complex and high‐scoring combinations thereof.
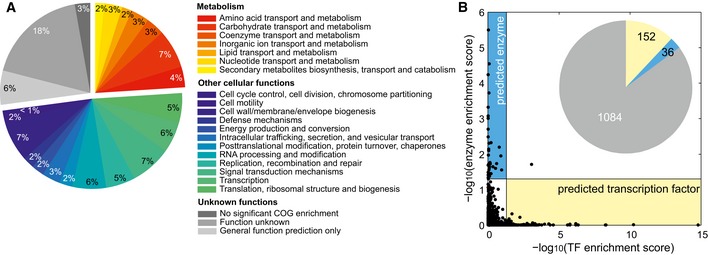
Enrichment of metabolic functions (defined by Clusters of Orthologous Groups, COG) for each y‐gene based on genes of known function with similar metabolome profiles, as determined by CLR.
Mutually exclusive function prediction of orphan genes as either enzymes or transcription factors (TF). The inset represents the number of genes predicted to be TFs (yellow), enzymes (blue), or neither (gray). One gene was predicted to be both a TF and an enzyme.
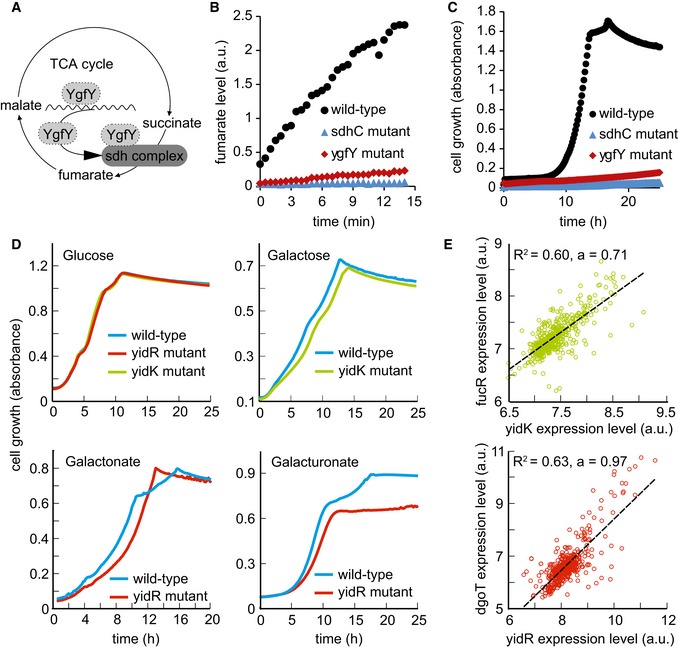
Potential functions of YgfY: transcriptional, post‐translational, or complex‐related modulator of succinate dehydrogenase activity.
Succinate dehydrogenase activity assay in cell lysates of wild‐type strain, sdhC, and ygfY mutants. Fumarate formation from supplied succinate was followed over time by mass spectrometry. Data are shown as mean and standard deviation of three replicates.
Growth defect of ygfY mutant on succinate minimal medium in comparison with wild‐type strain and sdhC mutant. Data are shown as mean and standard deviation of two replicates.
Growth defect of yidR and yidK mutants on mineral salt medium with the indicated carbon source in comparison with wild‐type strain. Solid line indicates mean from at least two replicates.
Correlation of expression levels over all 907 experiments in the M3D database. R 2 indicates goodness of a linear fit to the data and the strength of the correlation. FucR is a transcriptional activator of operons involved in fucose metabolism, and DgoT is a putative galactonate transporter.
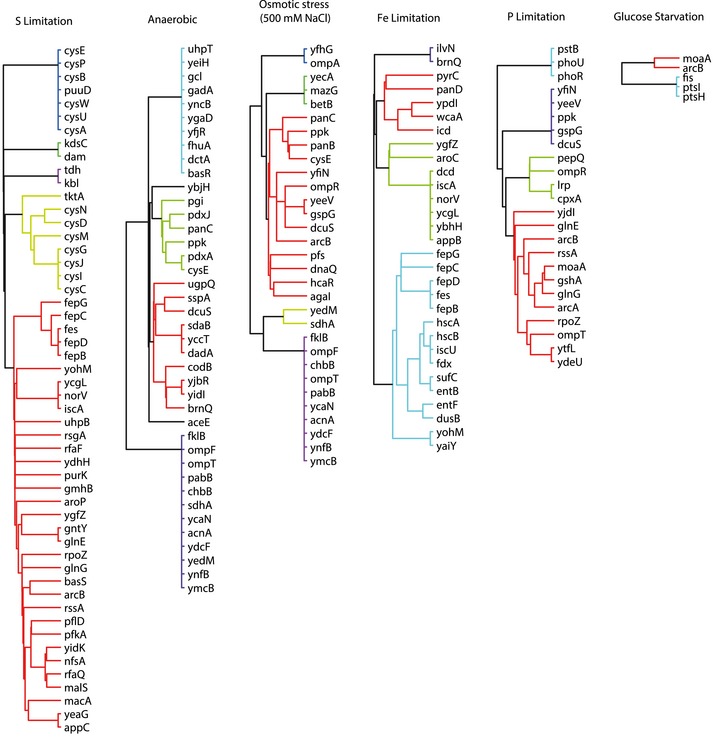
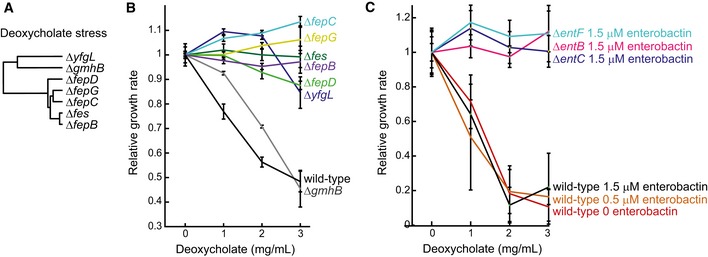
Dendrogram representing genes with significant overlap of differential metabolites in the respective knockout and during growth in the presence of 10 mg/ml deoxycholate in wild‐type Escherichia coli. Genes are hierarchically clustered based on their topological distance assessed by the minimum number of connecting reactions in the metabolic network.
Relative growth rates of wild‐type E. coli and deletion mutants in glucose minimal medium supplemented with casein hydrolysate and deoxycholate. Error bars represent standard deviations from three biological replicates.
Relative growth rates of wild‐type E. coli and enterobactin biosynthesis mutants in glucose minimal medium supplemented with enterobactin and deoxycholate. Error bars represent standard deviations from three biological replicates.
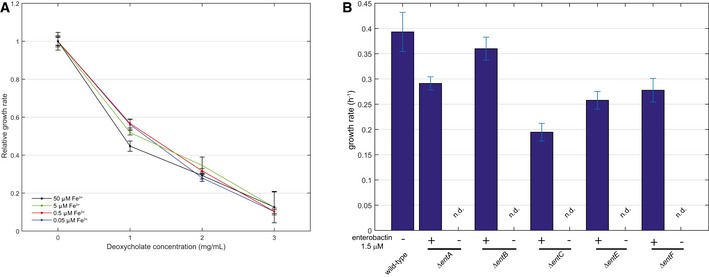
Escherichia coli wild‐type strain was grown on minimal medium casein hydrolysate with iron concentrations ranging from 0.05 up to 50 μM in the presence of 0, 1, 2, or 3 mg/ml deoxycholate. Relative growth rates were calculated during exponential growth and normalized to the 50 μM iron condition. Error bars represent standard deviations from three biological replicates.
Escherichia coli wild‐type strain and deletion mutants in enterobactin biosynthesis were grown on minimal medium without amino acids and with 1.5 μM (indicated with +) or without (indicated with −) enterobactin in the absence of deoxycholate. Maximum average growth rates with standard deviations during exponential growth phase were calculated from triplicate cultivations.
References
-
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, Prinz J, St Onge RP, VanderSluis B, Makhnevych T, Vizeacoumar FJ, Alizadeh S, Bahr S, Brost RL, Chen Y, Cokol M et al (2010) The genetic landscape of a cell. Science 327: 425–431 - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
