

Single-Cell Genomics: Approaches and Utility in Immunology
- PMID: 28094102
- PMCID: PMC5479322
- DOI: 10.1016/j.it.2016.12.001
Single-Cell Genomics: Approaches and Utility in Immunology
Abstract
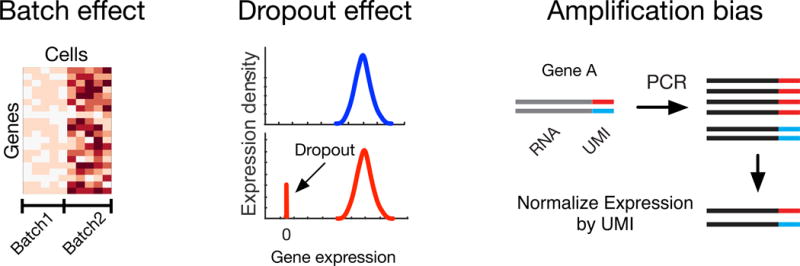
Single-cell genomics offers powerful tools for studying immune cells, which make it possible to observe rare and intermediate cell states that cannot be resolved at the population level. Advances in computer science and single-cell sequencing technology have created a data-driven revolution in immunology. The challenge for immunologists is to harness computing and turn an avalanche of quantitative data into meaningful discovery of immunological principles, predictive models, and strategies for therapeutics. Here, we review the current literature on computational analysis of single-cell RNA-sequencing data and discuss underlying assumptions, methods, and applications in immunology, and highlight important directions for future research.
Keywords: dimensionality reduction; immune repertoire; single-cell RNA-sequencing; visualization.
Copyright © 2016 Elsevier Ltd. All rights reserved.
Figures

References
-
- Tang F, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Meth. 2009;6:377–382. - PubMed
-
- Saeys Y, et al. Computational flow cytometry: helping to make sense of high-dimensional immunology data. Nat Rev Immunol. 2016;16:449–462. - PubMed
-
- Fulwyler MJ. Electronic Separation of Biological Cells by Volume. Science. 1965;150:910–911. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
