

Detection of a Potential New Bartonella Species "Candidatus Bartonella rondoniensis" in Human Biting Kissing Bugs (Reduviidae; Triatominae)
- PMID: 28095503
- PMCID: PMC5271407
- DOI: 10.1371/journal.pntd.0005297
Detection of a Potential New Bartonella Species "Candidatus Bartonella rondoniensis" in Human Biting Kissing Bugs (Reduviidae; Triatominae)
Abstract
Background: Among the Reduviidae family, triatomines are giant blood-sucking bugs. They are well known in Central and South America where they transmit Trypanosoma cruzi to mammals, including humans, through their feces. This parasitic protozoan is the causative agent of Chagas disease, a major public health issue in endemic areas. Because of the medical and economic impact of Chagas disease, the presence of other arthropod-borne pathogens in triatomines was rarely investigated.
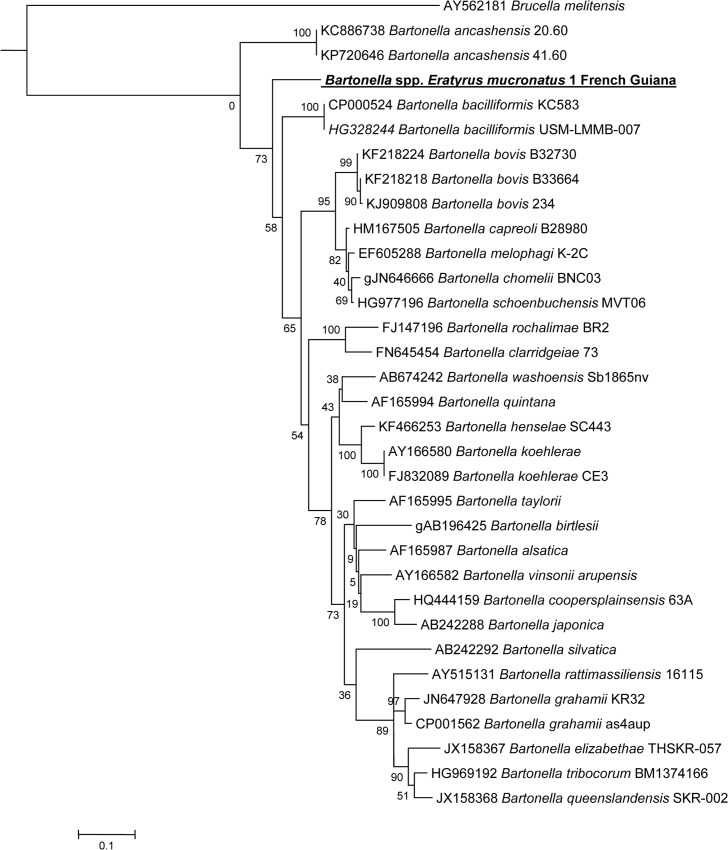
Methodology/principal findings: In this study, seven triatomines species involved in the transmission of T. cruzi were molecularly screened for the presence of known pathogens generally associated with arthropods, such as Rickettsia, Bartonella, Anaplasmataceae, Borrelia species and Coxiella burnetii. Of all included triatomine species, only Eratyrus mucronatus specimens tested positive for Bartonella species for 56% of tested samples. A new genotype of Bartonella spp. was detected in 13/23 Eratyrus mucronatus specimens, an important vector of T. cruzi to humans. This bacterium was further characterized by sequencing fragments of the ftsZ, gltA and rpoB genes. Depending on the targeted gene, this agent shares 84% to 91% of identity with B. bacilliformis, the agent of Carrion's disease, a deadly sandfly-borne infectious disease endemic in South America. It is also closely related to animal pathogens such as B. bovis and B. chomelii.
Conclusions: As E. mucronatus is an invasive species that occasionally feeds on humans, the presence of potentially pathogenic Bartonella-infected bugs could present another risk for human health, along with the T. cruzi issue.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures




Similar articles
-
Triatoma rubrofasciata as a potential vector for bartonellosis.Emerg Microbes Infect. 2025 Dec;14(1):2494291. doi: 10.1080/22221751.2025.2494291. Epub 2025 May 2. Emerg Microbes Infect. 2025. PMID: 40231453 Free PMC article.
-
Detection of Bartonella in kissing bugs Triatoma rubrofasciata collected from Huizhou City, South China.New Microbes New Infect. 2023 Sep 1;54:101170. doi: 10.1016/j.nmni.2023.101170. eCollection 2023 Sep. New Microbes New Infect. 2023. PMID: 37692291 Free PMC article.
-
Triatomines: Trypanosomatids, Bacteria, and Viruses Potential Vectors?Front Cell Infect Microbiol. 2018 Nov 16;8:405. doi: 10.3389/fcimb.2018.00405. eCollection 2018. Front Cell Infect Microbiol. 2018. PMID: 30505806 Free PMC article. Review.
-
Detection of Trypansoma cruzi in Kissing Bugs (Hemiptera: Reduviidae: Triatominae) Collected Across Oklahoma.J Med Entomol. 2022 Mar 16;59(2):675-680. doi: 10.1093/jme/tjab211. J Med Entomol. 2022. PMID: 34993549
-
Evolution, Systematics, and Biogeography of the Triatominae, Vectors of Chagas Disease.Adv Parasitol. 2018;99:265-344. doi: 10.1016/bs.apar.2017.12.002. Adv Parasitol. 2018. PMID: 29530308 Review.
Cited by
-
Distribution, genetic characteristics and public health implications of Triatoma rubrofasciata, the vector of Chagas disease in Guangxi, China.Parasit Vectors. 2020 Jan 20;13(1):33. doi: 10.1186/s13071-020-3903-z. Parasit Vectors. 2020. PMID: 31959216 Free PMC article.
-
Global fingerprint of humans on the distribution of Bartonella bacteria in mammals.PLoS Negl Trop Dis. 2018 Nov 15;12(11):e0006865. doi: 10.1371/journal.pntd.0006865. eCollection 2018 Nov. PLoS Negl Trop Dis. 2018. PMID: 30439961 Free PMC article.
-
Triatoma rubrofasciata as a potential vector for bartonellosis.Emerg Microbes Infect. 2025 Dec;14(1):2494291. doi: 10.1080/22221751.2025.2494291. Epub 2025 May 2. Emerg Microbes Infect. 2025. PMID: 40231453 Free PMC article.
-
Detection of Bartonella in kissing bugs Triatoma rubrofasciata collected from Huizhou City, South China.New Microbes New Infect. 2023 Sep 1;54:101170. doi: 10.1016/j.nmni.2023.101170. eCollection 2023 Sep. New Microbes New Infect. 2023. PMID: 37692291 Free PMC article.
-
Using MALDI-TOF mass spectrometry to identify ticks collected on domestic and wild animals from the Democratic Republic of the Congo.Exp Appl Acarol. 2021 Jul;84(3):637-657. doi: 10.1007/s10493-021-00629-z. Epub 2021 Jun 19. Exp Appl Acarol. 2021. PMID: 34146230 Free PMC article.
References
-
- Schofield CJ, Galvão C. Classification, evolution, and species groups within the Triatominae. Acta Trop. 2009;110: 88–100. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
