

Prion-Like Characteristics of Polyglutamine-Containing Proteins
- PMID: 28096245
- PMCID: PMC5793740
- DOI: 10.1101/cshperspect.a024257
Prion-Like Characteristics of Polyglutamine-Containing Proteins
Abstract
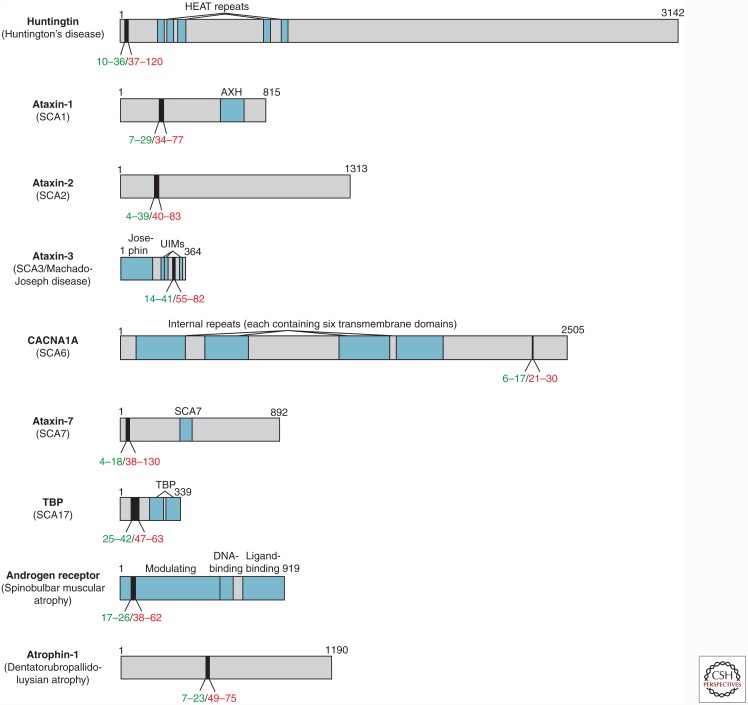
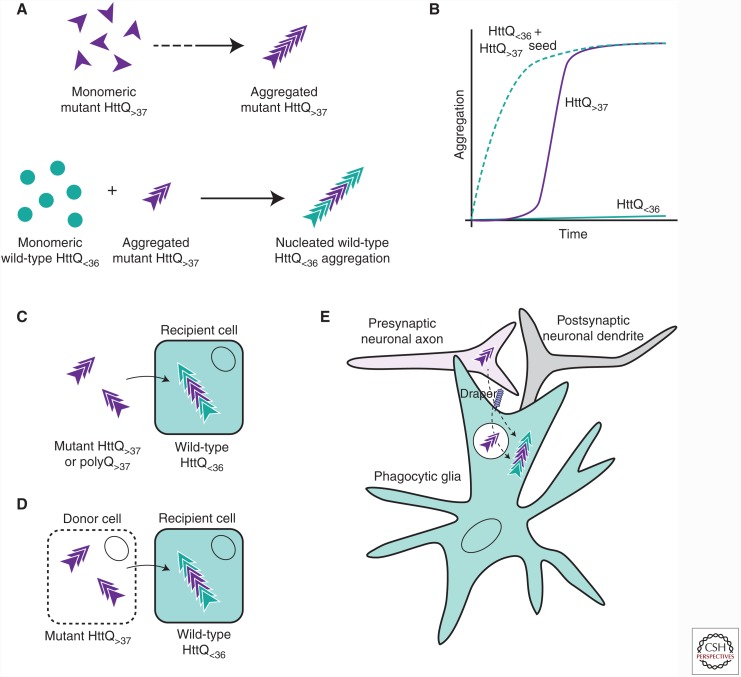
Transmissible spongiform encephalopathies are infectious neurodegenerative diseases caused by the conversion of prion protein (PrP) into a self-replicating conformation that spreads via templated conversion of natively folded PrP molecules within or between cells. Recent studies provide compelling evidence that prion-like behavior is a general property of most protein aggregates associated with neurodegenerative diseases. Many of these disorders are associated with spontaneous protein aggregation, but genetic mutations can increase the aggregation propensity of specific proteins, including expansion of polyglutamine (polyQ) tracts, which is causative of nine inherited neurodegenerative diseases. Aggregates formed by polyQ-expanded huntingtin (Htt) in Huntington's disease can transfer between cells and seed the aggregation of cytoplasmic wild-type Htt in a prion-like manner. Additionally, prion-like properties of glutamine-rich proteins underlie nonpathological processes in yeast and higher eukaryotes. Here, we review current evidence supporting prion-like characteristics of polyQ and glutamine-rich proteins.
Copyright © 2018 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
-
- Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H, Parhizkar S, Ward MA, Cavallini A, Jackson S, et al. 2014. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: The pattern of spread is determined by connectivity, not proximity. Acta Neuropathol 127: 667–683. - PMC - PubMed
-
- Alper T, Cramp WA, Haig DA, Clarke MC. 1967. Does the agent of scrapie replicate without nucleic acid? Nature 214: 764–766. - PubMed
-
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. 2004. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431: 805–810. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials