

Order Matters: The Order of Somatic Mutations Influences Cancer Evolution
- PMID: 28096247
- PMCID: PMC5378012
- DOI: 10.1101/cshperspect.a027060
Order Matters: The Order of Somatic Mutations Influences Cancer Evolution
Abstract
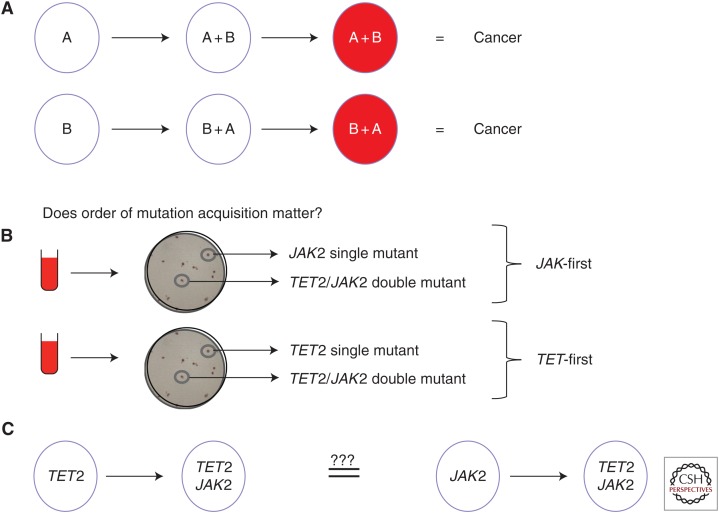
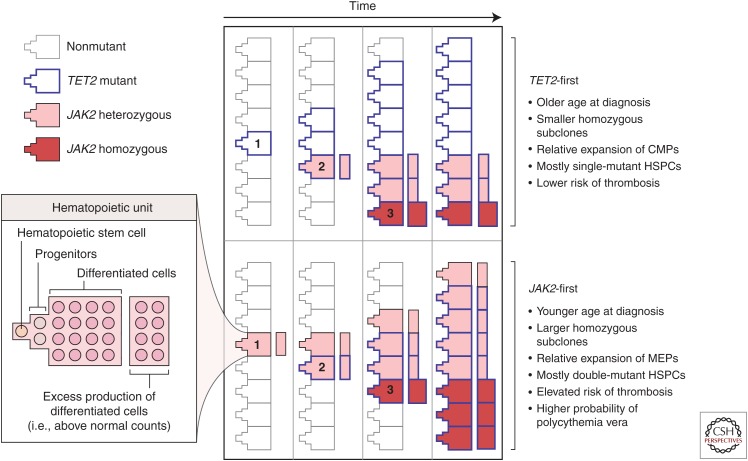
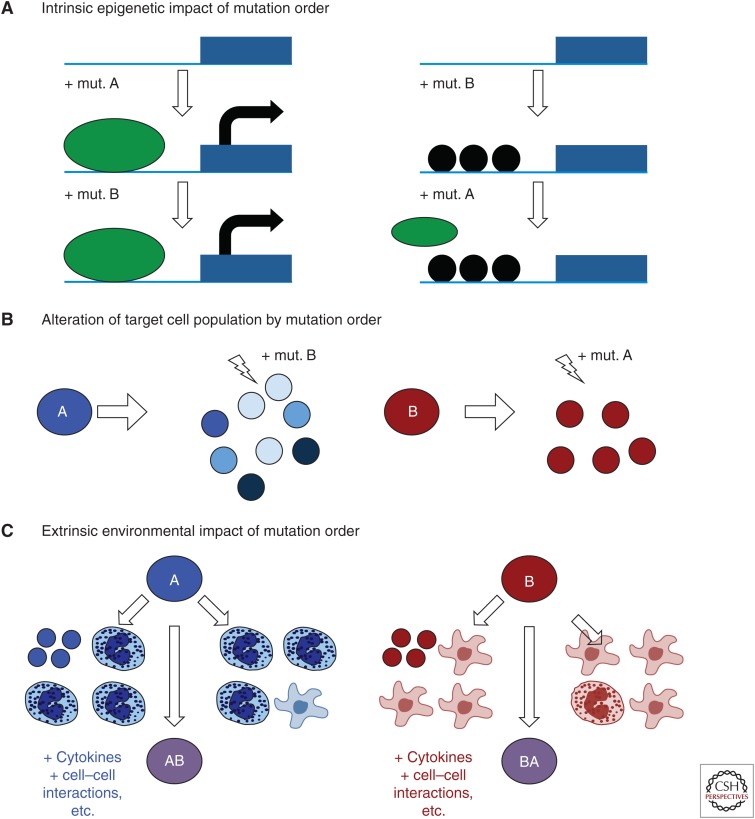
Cancers evolve as a consequence of multiple somatic lesions, with competition between subclones and sequential subclonal evolution. Some driver mutations arise either early or late in the evolution of different individual tumors, suggesting that the final malignant properties of a subclone reflect the sum of mutations acquired rather than the order in which they arose. However, very little is known about the cellular consequences of altering the order in which mutations are acquired. Recent studies of human myeloproliferative neoplasms show that the order in which individual mutations are acquired has a dramatic impact on the cell biological and molecular properties of tumor-initiating cells. Differences in clinical presentation, complications, and response to targeted therapy were all observed and implicate mutation order as an important player in cancer biology. These observations represent the first demonstration that the order of mutation acquisition influences stem and progenitor cell behavior and clonal evolution in any cancer. Thus far, the impact of different mutation orders has only been studied in hematological malignancies, and analogous studies of solid cancers are now required.
Copyright © 2017 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
Similar articles
-
Principles of Reconstructing the Subclonal Architecture of Cancers.Cold Spring Harb Perspect Med. 2017 Aug 1;7(8):a026625. doi: 10.1101/cshperspect.a026625. Cold Spring Harb Perspect Med. 2017. PMID: 28270531 Free PMC article. Review.
-
Somatic evolutionary timings of driver mutations.BMC Cancer. 2018 Jan 18;18(1):85. doi: 10.1186/s12885-017-3977-y. BMC Cancer. 2018. PMID: 29347918 Free PMC article.
-
Cancer heterogeneity: converting a limitation into a source of biologic information.J Transl Med. 2017 Sep 8;15(1):190. doi: 10.1186/s12967-017-1290-9. J Transl Med. 2017. PMID: 28886708 Free PMC article. Review.
-
SubClonal Hierarchy Inference from Somatic Mutations: Automatic Reconstruction of Cancer Evolutionary Trees from Multi-region Next Generation Sequencing.PLoS Comput Biol. 2015 Oct 5;11(10):e1004416. doi: 10.1371/journal.pcbi.1004416. eCollection 2015 Oct. PLoS Comput Biol. 2015. PMID: 26436540 Free PMC article.
-
Rare subclonal sequencing of breast cancers indicates putative metastatic driver mutations are predominately acquired after dissemination.Genome Med. 2024 Feb 6;16(1):26. doi: 10.1186/s13073-024-01293-9. Genome Med. 2024. PMID: 38321573 Free PMC article.
Cited by
-
Genetic Determinants of Somatic Selection of Mutational Processes in 3,566 Human Cancers.Cancer Res. 2021 Aug 15;81(16):4205-4217. doi: 10.1158/0008-5472.CAN-21-0086. Epub 2021 Jul 2. Cancer Res. 2021. PMID: 34215622 Free PMC article.
-
The dark side of stemness - the role of hematopoietic stem cells in development of blood malignancies.Front Oncol. 2024 Feb 19;14:1308709. doi: 10.3389/fonc.2024.1308709. eCollection 2024. Front Oncol. 2024. PMID: 38440231 Free PMC article. Review.
-
Trade-offs, trade-ups, and high mutational parallelism underlie microbial adaptation during extreme cycles of feast and famine.Curr Biol. 2024 Apr 8;34(7):1403-1413.e5. doi: 10.1016/j.cub.2024.02.040. Epub 2024 Mar 8. Curr Biol. 2024. PMID: 38460514 Free PMC article.
-
Generation of autochthonous mouse models of clear cell renal cell carcinoma: mouse models of renal cell carcinoma.Exp Mol Med. 2018 Apr 13;50(4):1-10. doi: 10.1038/s12276-018-0059-4. Exp Mol Med. 2018. PMID: 29651023 Free PMC article. Review.
-
Exploiting Single-Cell Tools in Gene and Cell Therapy.Front Immunol. 2021 Jul 12;12:702636. doi: 10.3389/fimmu.2021.702636. eCollection 2021. Front Immunol. 2021. PMID: 34322133 Free PMC article. Review.
References
-
- Alderton GK. 2015. Tumour microenvironment: Driving relapse. Nat Rev Cancer 15: 195. - PubMed
-
- Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, Kempski H, Moorman AV, Titley I, Swansbury J, et al. 2011. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 469: 356–361. - PubMed
-
- Balmain A. 2001. Cancer genetics: From Boveri and Mendel to microarrays. Nat Rev Cancer 1: 77–82. - PubMed
-
- Basheer F, Huntly BJ. 2015. BET bromodomain inhibitors in leukemia. Exp Hematol 43: 718–731. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources