

Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis
- PMID: 28096490
- PMCID: PMC5241402
- DOI: 10.1128/mBio.02133-16
Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis
Abstract
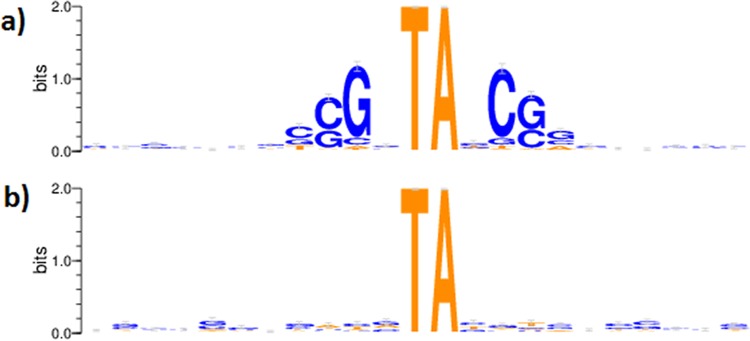
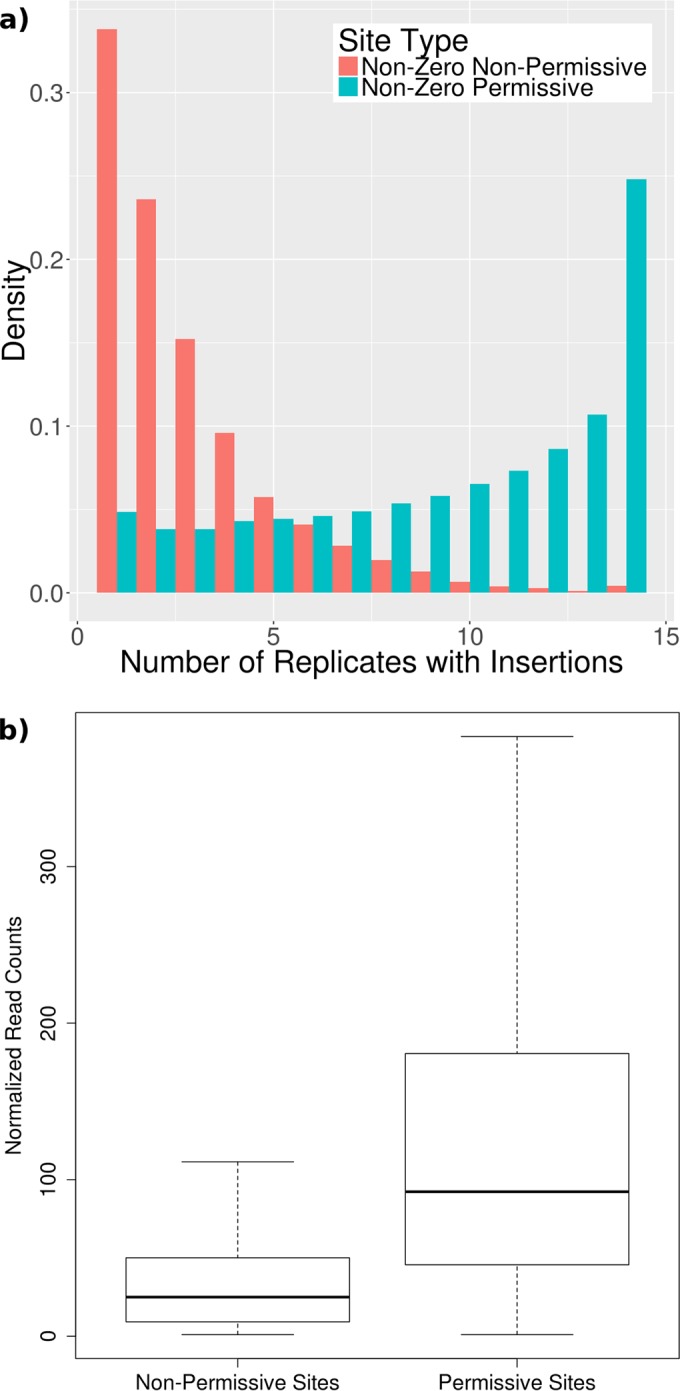
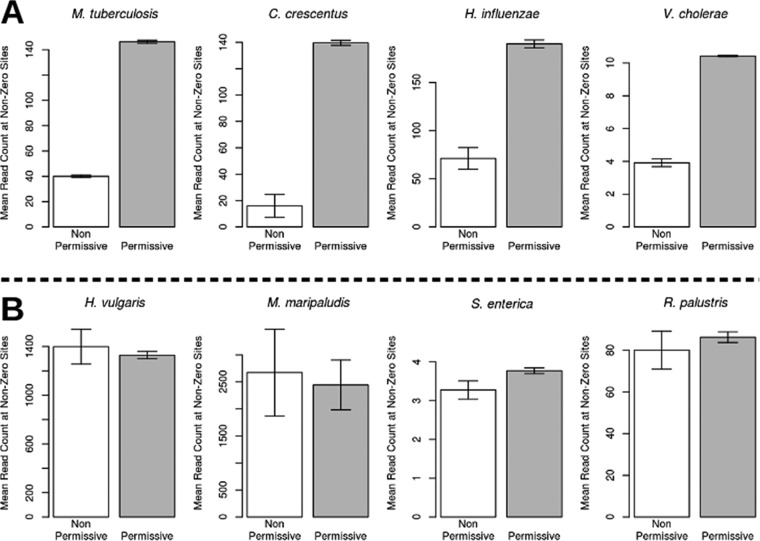
For decades, identifying the regions of a bacterial chromosome that are necessary for viability has relied on mapping integration sites in libraries of random transposon mutants to find loci that are unable to sustain insertion. To date, these studies have analyzed subsaturated libraries, necessitating the application of statistical methods to estimate the likelihood that a gap in transposon coverage is the result of biological selection and not the stochasticity of insertion. As a result, the essentiality of many genomic features, particularly small ones, could not be reliably assessed. We sought to overcome this limitation by creating a completely saturated transposon library in Mycobacterium tuberculosis In assessing the composition of this highly saturated library by deep sequencing, we discovered that a previously unknown sequence bias of the Himar1 element rendered approximately 9% of potential TA dinucleotide insertion sites less permissible for insertion. We used a hidden Markov model of essentiality that accounted for this unanticipated bias, allowing us to confidently evaluate the essentiality of features that contained as few as 2 TA sites, including open reading frames (ORF), experimentally identified noncoding RNAs, methylation sites, and promoters. In addition, several essential regions that did not correspond to known features were identified, suggesting uncharacterized functions that are necessary for growth. This work provides an authoritative catalog of essential regions of the M. tuberculosis genome and a statistical framework for applying saturating mutagenesis to other bacteria.
Importance: Sequencing of transposon-insertion mutant libraries has become a widely used tool for probing the functions of genes under various conditions. The Himar1 transposon is generally believed to insert with equal probabilities at all TA dinucleotides, and therefore its absence in a mutant library is taken to indicate biological selection against the corresponding mutant. Through sequencing of a saturated Himar1 library, we found evidence that TA dinucleotides are not equally permissive for insertion. The insertion bias was observed in multiple prokaryotes and influences the statistical interpretation of transposon insertion (TnSeq) data and characterization of essential genomic regions. Using these insights, we analyzed a fully saturated TnSeq library for M. tuberculosis, enabling us to generate a comprehensive catalog of in vitro essentiality, including ORFs smaller than those found in any previous study, small (noncoding) RNAs (sRNAs), promoters, and other genomic features.
Copyright © 2017 DeJesus et al.
Figures
References
-
- Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, Chun-Rong L, Guenthner D, Bovee D, Olson MV, Manoil C. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 100:14339–14344. doi:10.1073/pnas.2036282100. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
