

Diagnosis for choroideremia in a large Chinese pedigree by next‑generation sequencing (NGS) and non‑invasive prenatal testing (NIPT)
- PMID: 28098911
- PMCID: PMC5367376
- DOI: 10.3892/mmr.2017.6119
Diagnosis for choroideremia in a large Chinese pedigree by next‑generation sequencing (NGS) and non‑invasive prenatal testing (NIPT)
Abstract
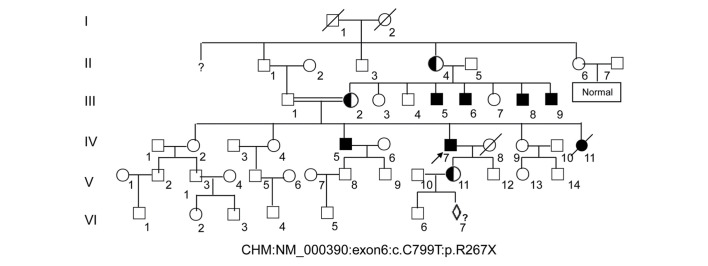
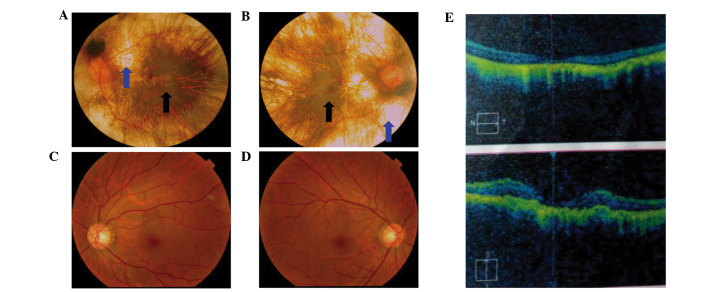
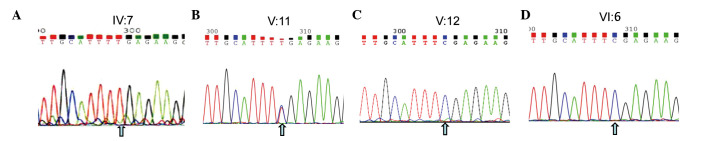
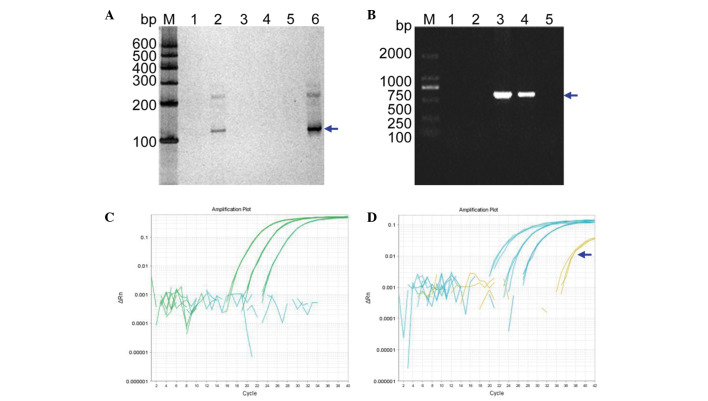
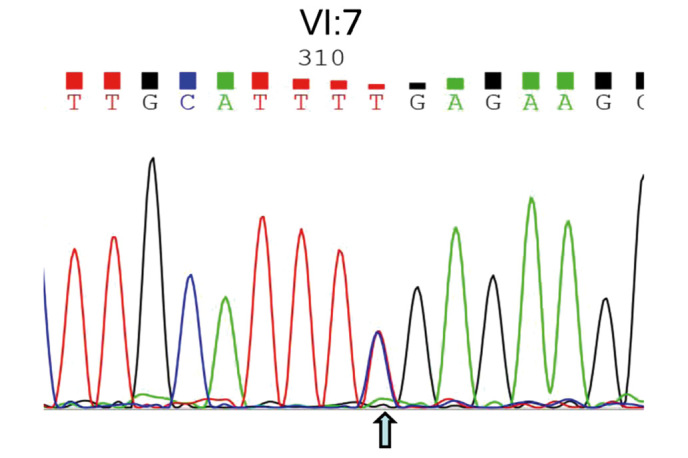
To develop an effective strategy to isolate and use cell‑free fetal DNA (cffDNA) for the combined use of next‑generation sequencing (NGS) for diagnosing choroideremia and non‑invasive prenatal testing (NIPT) for Y chromosome determination, a large Chinese family with an X‑linked recessive disease, choroideremia, was recruited. Cell‑free DNA was extracted from maternal plasma, and SRY polymerase chain reaction amplification was performed using NIPT. Sanger sequencing was subsequently used for fetal amniotic fluid DNA verification. A nonsense mutation (c.C799T:p.R267X) of the CHM gene on the X chromosome of the proband (IV:7) and another 5 males with choroideremia were detected, while 3 female carriers with no symptoms were also identified. The fetus (VI:7) was identified as female from the cffDNA, and the same heterozygous nonsense mutation present in her mother was also confirmed. At one and a half years of age, the female baby did not present with any associated symptoms of choroideremia. Therefore, cffDNA was successfully used for the combined use of NGS for diagnosing choroideremia in a large Chinese pedigree, and NIPT for Y chromosome determination. This approach should result in a markedly increased use of prenatal diagnosis and improvement, and more sophisticated clinical management of diseases in China and other developing countries. The establishment of a highly accurate method for prenatal gene diagnosis will allow for more reliable gene diagnosis, improved genetic counseling, and personalized clinical management of our patients.
Figures
References
-
- Li S, Guan L, Fang S, Jiang H, Xiao X, Yang J, Wang P, Yin Y, Guo X, Wang J, et al. Exome sequencing reveals CHM mutations in six families with atypical choroideremia initially diagnosed as retinitis pigmentosa. Int J Mol Med. 2014;34:573–577. - PubMed
-
- Esposito G, De Falco F, Tinto N, Testa F, Vitagliano L, Tandurella IC, Iannone L, Rossi S, Rinaldi E, Simonelli F, et al. Comprehensive mutation analysis (20 families) of the choroideremia gene reveals a missense variant that prevents the binding of REP1 with Rab geranylgeranyl transferase. Hum Mutat. 2011;32:1460–1469. doi: 10.1002/humu.21591. - DOI - PubMed
-
- Garcia-Hoyos M, Lorda-Sanchez I, Gómez-Garre P, Villaverde C, Cantalapiedra D, Bustamante A, Diego-Alvarez D, Vallespin E, Gallego-Merlo J, Trujillo MJ, et al. New type of mutations in three Spanish families with choroideremia. Invest Ophthalmol Vis Sci. 2008;49:1315–1321. doi: 10.1167/iovs.07-1169. - DOI - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
