

An Exome Sequencing Study to Assess the Role of Rare Genetic Variation in Pulmonary Fibrosis
- PMID: 28099038
- PMCID: PMC5519963
- DOI: 10.1164/rccm.201610-2088OC
An Exome Sequencing Study to Assess the Role of Rare Genetic Variation in Pulmonary Fibrosis
Abstract
Rationale: Idiopathic pulmonary fibrosis (IPF) is an increasingly recognized, often fatal lung disease of unknown etiology.
Objectives: The aim of this study was to use whole-exome sequencing to improve understanding of the genetic architecture of pulmonary fibrosis.
Methods: We performed a case-control exome-wide collapsing analysis including 262 unrelated individuals with pulmonary fibrosis clinically classified as IPF according to American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Association guidelines (81.3%), usual interstitial pneumonia secondary to autoimmune conditions (11.5%), or fibrosing nonspecific interstitial pneumonia (7.2%). The majority (87%) of case subjects reported no family history of pulmonary fibrosis.
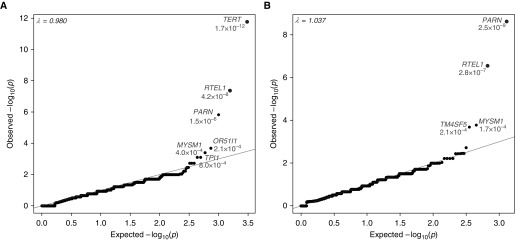
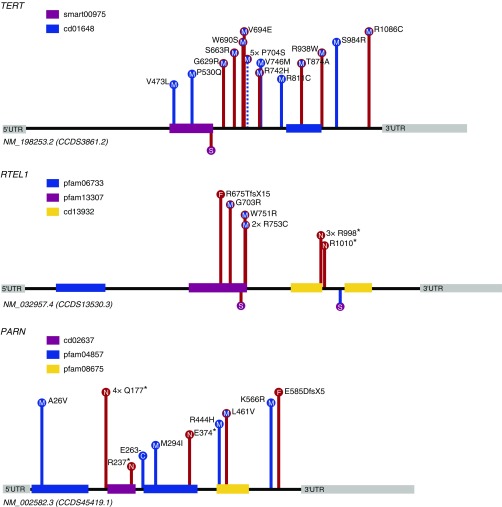
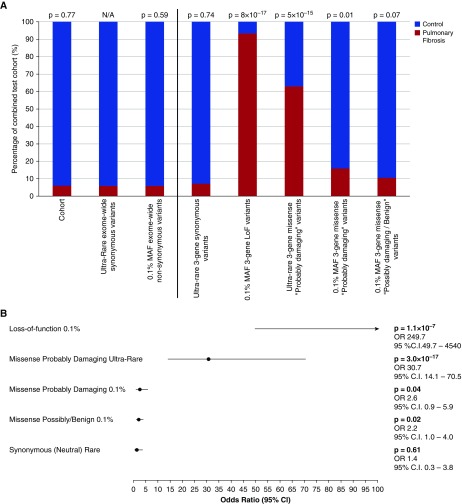
Measurements and main results: We searched 18,668 protein-coding genes for an excess of rare deleterious genetic variation using whole-exome sequence data from 262 case subjects with pulmonary fibrosis and 4,141 control subjects drawn from among a set of individuals of European ancestry. Comparing genetic variation across 18,668 protein-coding genes, we found a study-wide significant (P < 4.5 × 10-7) case enrichment of qualifying variants in TERT, RTEL1, and PARN. A model qualifying ultrarare, deleterious, nonsynonymous variants implicated TERT and RTEL1, and a model specifically qualifying loss-of-function variants implicated RTEL1 and PARN. A subanalysis of 186 case subjects with sporadic IPF confirmed TERT, RTEL1, and PARN as study-wide significant contributors to sporadic IPF. Collectively, 11.3% of case subjects with sporadic IPF carried a qualifying variant in one of these three genes compared with the 0.3% carrier rate observed among control subjects (odds ratio, 47.7; 95% confidence interval, 21.5-111.6; P = 5.5 × 10-22).
Conclusions: We identified TERT, RTEL1, and PARN-three telomere-related genes previously implicated in familial pulmonary fibrosis-as significant contributors to sporadic IPF. These results support the idea that telomere dysfunction is involved in IPF pathogenesis.
Keywords: collapsing analysis; exome sequencing; genetics; interstitial lung disease; pulmonary fibrosis.
Figures
Comment in
-
Whole-Exome Sequencing Insights into Adult Pulmonary Fibrosis. Repeating the Telomere Theme.Am J Respir Crit Care Med. 2017 Jul 1;196(1):7-9. doi: 10.1164/rccm.201701-0194ED. Am J Respir Crit Care Med. 2017. PMID: 28665198 Free PMC article. No abstract available.
References
-
- Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et al. ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. - PMC - PubMed
-
- Raghu G, Chen SY, Yeh WS, Maroni B, Li Q, Lee YC, Collard HR.Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001–11 Lancet Respir Med 20142566–572.[Published erratum appears in Lancet Respir Med 2014;2:e12.] - PubMed
-
- Steele MP, Schwartz DA. Molecular mechanisms in progressive idiopathic pulmonary fibrosis. Annu Rev Med. 2013;64:265–276. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources