

Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype
- PMID: 28100023
- PMCID: PMC8029341
- DOI: 10.1111/bpa.12486
Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype
Abstract
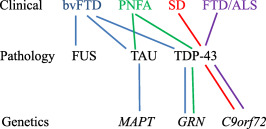
Frontotemporal Lobar Degeneration (FTLD) is a clinically, pathologically and genetically heterogeneous group of disorders that affect principally the frontal and temporal lobes of the brain. There are three major associated clinical syndromes, behavioral variant frontotemporal dementia (bvFTD), semantic dementia (SD) and progressive non-fluent aphasia (PNFA); three principal histologies, involving tau, TDP-43 and FUS proteins; and mutations in three major genes, MAPT, GRN and C9orf72, along with several other less common gene mutations. All three clinical syndromes can exist separately or in combination with Amyotrophic Lateral Sclerosis (ALS). SD is exclusively a TDP-43 proteinopathy, and PNFA may be so, with both showing tight clinical, histological and genetic inter-relationships. bvFTD is more of a challenge with overlapping histological and genetic features, involvement of any of the three aggregating proteins, and changes in any of the three major genes. However, when ALS is present, all cases show a clear histological phenotype with TDP-43 aggregated proteins, and familial forms are associated with expansions in C9orf72. TDP-43 and FUS are nuclear carrier proteins involved in the regulation of RNA metabolism, whereas tau protein - the product of MAPT - is responsible for the assembly/disassembly of microtubules, which are vital for intracellular transport. Mutations in TDP-43 and FUS genes are linked to clinical ALS rather than FTLD (with or without ALS), suggesting that clinical ALS may be a disorder of RNA metabolism. Conversely, the protein products of GRN and C9orf72, along with those of the other minor genes, appear to form part of the cellular protein degradation machinery. It is possible therefore that FTLD is a reflection of dysfunction within lysosomal/proteasomal systems resulting in failure to remove potentially neurotoxic (TDP-43 and tau) aggregates, which ultimately overwhelm capacity to function. Spread of aggregates along distinct pathways may account for the different clinical phenotypes, and patterns of progression of disease.
Keywords: clinical phenotypes; frontotemporal lobar degeneration; genetics; neuropathology; pathogenesis.
© 2017 International Society of Neuropathology.
Conflict of interest statement
The authors have no Conflicts of Interest to declare.
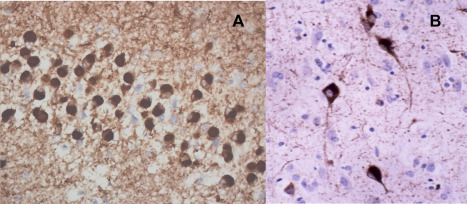
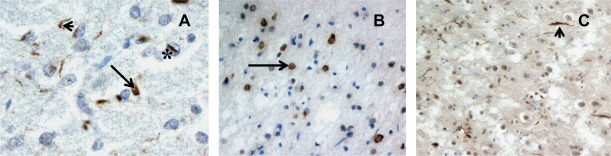
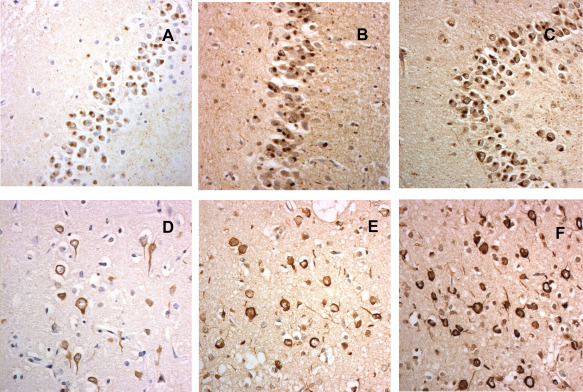
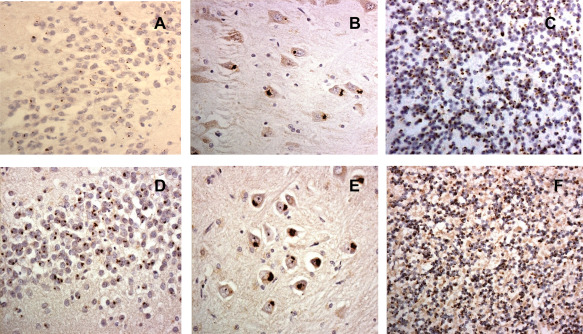
Figures
References
-
- Acosta‐Cabronero J, Patterson K, Fryer TD, Hodges JR, Pengas G, Williams GB et al (2011) Atrophy, hypometabolism and white matter abnormalities in semantic dementia tell a coherent story. Brain 134:2025–2035. - PubMed
-
- Agosta F, Galantucci S, Canu E, Cappa SF, Magnani G, Franceschi M et al (2013) Disruption of structural connectivity along the dorsal and ventral language pathways in patients with nonfluent and semantic variant primary progressive aphasia: A DT MRI study and a literature review. Brain Lang 127:157–166. - PubMed
-
- Armstrong RA (2016) Survival in the pre‐senile dementia frontotemporal lobar degeneration with TDP‐43 proteinopathy: Effects of genetic, demographic and neuropathological variables. Folia Neuropathol 54:137–148. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
