

SMITE: an R/Bioconductor package that identifies network modules by integrating genomic and epigenomic information
- PMID: 28100166
- PMCID: PMC5242055
- DOI: 10.1186/s12859-017-1477-3
SMITE: an R/Bioconductor package that identifies network modules by integrating genomic and epigenomic information
Abstract
Background: The molecular assays that test gene expression, transcriptional, and epigenetic regulation are increasingly diverse and numerous. The information generated by each type of assay individually gives an insight into the state of the cells tested. What should be possible is to add the information derived from separate, complementary assays to gain higher-confidence insights into cellular states. At present, the analysis of multi-dimensional, massive genome-wide data requires an initial pruning step to create manageable subsets of observations that are then used for integration, which decreases the sizes of the intersecting data sets and the potential for biological insights. Our Significance-based Modules Integrating the Transcriptome and Epigenome (SMITE) approach was developed to integrate transcriptional and epigenetic regulatory data without a loss of resolution.
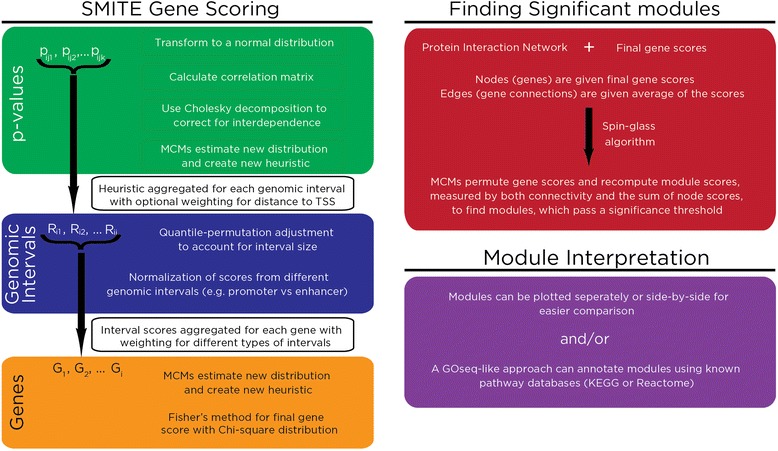
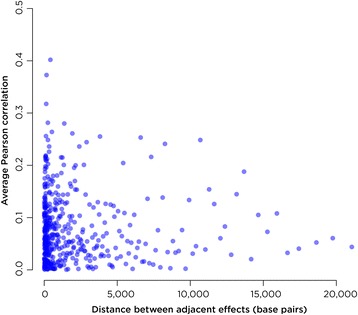
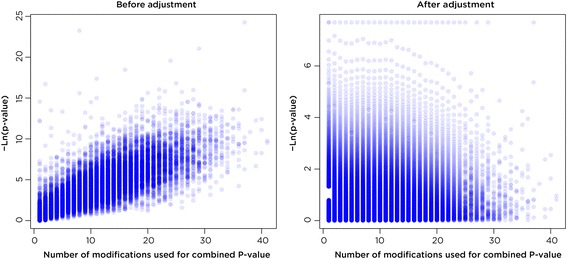
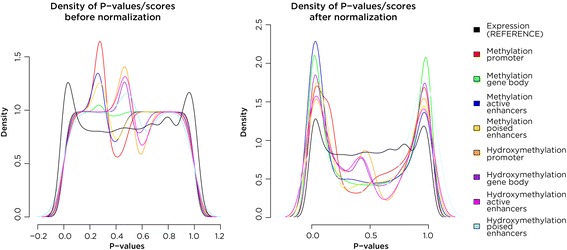
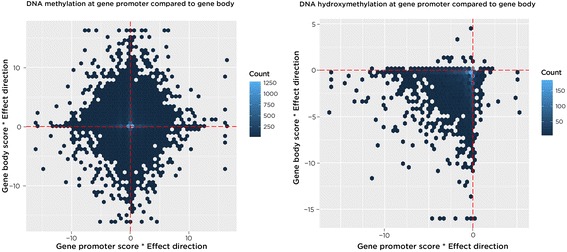
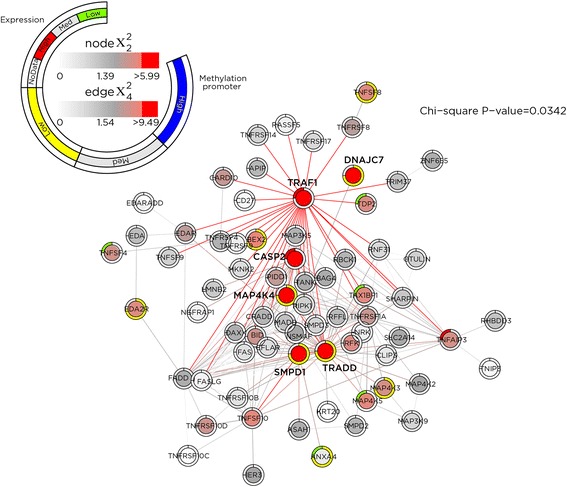
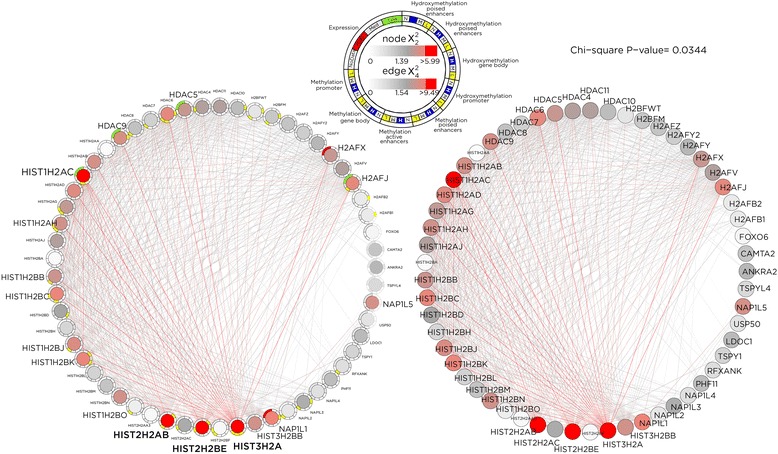
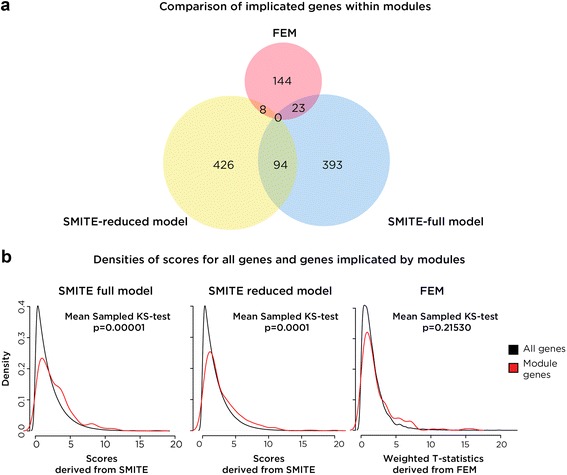
Results: SMITE combines p-values by accounting for the correlation between non-independent values within data sets, allowing genes and gene modules in an interaction network to be assigned significance values. The contribution of each type of genomic data can be weighted, permitting integration of individually under-powered data sets, increasing the overall ability to detect effects within modules of genes. We apply SMITE to a complex genomic data set including the epigenomic and transcriptomic effects of Toxoplasma gondii infection on human host cells and demonstrate that SMITE is able to identify novel subnetworks of dysregulated genes. Additionally, we show that SMITE outperforms Functional Epigenetic Modules (FEM), the current paradigm of using the spin-glass algorithm to integrate gene expression and epigenetic data.
Conclusions: SMITE represents a flexible, scalable tool that allows integration of transcriptional and epigenetic regulatory data from genome-wide assays to boost confidence in finding gene modules reflecting altered cellular states.
Keywords: Bioinformatics; Epigenetic; Gene expression; Genomic; Interaction network; Modules.
Figures
References
-
- Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, Rando OJ, Birney E, Myers RM, Noble WS, Snyder M, Weng Z. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798–812. doi: 10.1101/gr.139105.112. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
